


 收藏
收藏 已收藏
已收藏英文名称 :progressive familial intrahepatic cholestasis-6
进行性家族性肝内胆汁淤积症6型(progressive familial intrahepatic cholestasis-6,PFIC-6)是近期发现的家族性遗传性肝内胆汁淤积症,由编码Vb型肌球蛋白的MYO5B基因突变导致,该病属于常染色体隐性遗传。王建设教授团队在1/5的儿童不明原因低GGT胆汁淤积患者发现了MYO5B双突变,但目前国内外尚无关于本病的大样本报道和流行病学数据。
MYO5B基因位于染色体18q21.1,含有40个外显子和39个内含子,基因组全长372kb,编码由1848个氨基酸组成的Ⅴb型肌球蛋白。Ⅴ型肌球蛋白家族在组织细胞中广泛表达。MYO5B含actin结合位点,介导actin依赖的物质运输;在极性细胞中,MYO5B与不同的Rab家族蛋白调控细胞内不同的囊泡再循环通路(recycling pathway),参与高尔基体-细胞膜之间的物质循环、物质定向转运,以及极性细胞极性面的形成。MYO5B与Rab8A或Rab11A的相互作用消失均会使得肠上皮细胞中原本定位于刷状缘的囊泡无法正常定位,正常极性结构不能形成,导致氯离子分泌通道表达受损,被认为是MYO5B缺陷引起腹泻的主要致病机制。以往有报道部分存活时间较长的腹泻表型病例会伴发胆汁淤积,曾被认为是肠外营养的副作用。但最近研究发现MYO5B突变继发BSEP缺陷可能是其导致胆汁淤积症的重要致病机制。患者临床表现为与BSEP缺陷病相似的全谱系低GGT胆汁淤积症,血浆胆汁酸谱改变也与BSEP缺陷病非常类似。独立胆汁淤积表型的MYO5B突变病例BSEP不仅定位出现异常,表达也发生明显减少或缺如。
目前,已发现导致胆汁淤积MYO5B基因突变包括无义突变(c.1021C>T,p.Q341X;c.3046C>T,p.R1016X)、错义突变(c.1604G>A,p.S535N)及c.2470C>T,p.R824C、c.1136G>C,p.R379P、c.1135C>T,p.R379P。MYO5B基因缺陷者表型差异可能和突变严重程度有关,腹泻表型病例携带突变预测影响MYO5B-RAB11A相互作用区域的比例显著高于独立胆汁淤积表型病例。
MYO5B基因缺陷胆汁淤积病例肝组织病理表现为肝细胞排列紊乱、巨细胞样变,伴细胞内及毛细胆管内胆汁淤积。CK7及CK19染色提示轻度胆管增生;MYO5B免疫组化染色显示MYO5B阳性颗粒明显增多、增大,呈弥漫性粗颗粒改变;可存在不同程度的BSEP表达减少。病理表现见彩图1。
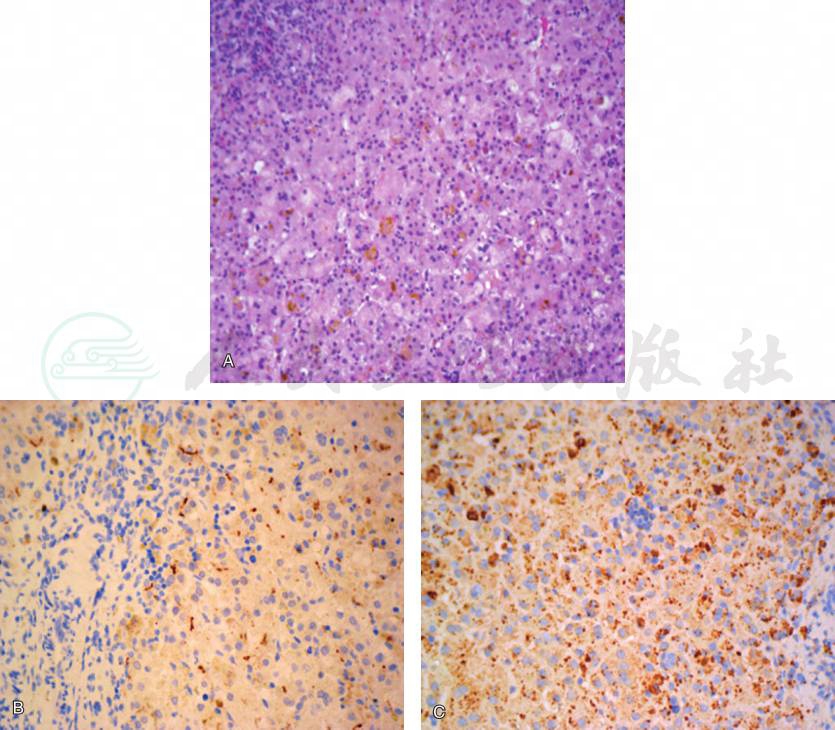
彩图1MYO5B基因缺陷胆汁淤积病例肝组织病理表现
A.肝组织HE染色巨细胞样变,伴细胞内及毛细胆管内胆汁淤积;B.免疫组化BSEP染色表达减少;C.免疫组化MYO5B阳性颗粒明显增多、增大,呈弥漫性粗颗粒改变
引自:遗传代谢病防治理论与实践.第1版.ISBN:978-7-117-34392-3.主编:
血生化提示总胆红素、直接胆红素升高,转氨酶轻度升高,GGT正常,总胆汁酸升高;血糖、血氨、甲胎蛋白在正常范围内;如果严重腹泻病患者可有代谢性酸中毒、电解质紊乱、多种维生素缺乏。
与其他家族性胆汁淤积症一样,PFIC-6必须补充脂溶性维生素。富含中链脂肪酸营养补充,可保证能量。抗组胺药及熊去氧胆酸对瘙痒的作用有限,考来烯胺可能有一定的作用。









