


 收藏
收藏 已收藏
已收藏英文名称 :creatine deficiency syndrome
肌酸缺乏综合征(creatine deficiency syndrome)是一组先天性肌酸合成及转运异常疾病,主要包括精氨酸:甘氨酸转脒酶缺乏症[arginine:glycine amidinotransferase(AGAT)deficiency]、胍基乙酸甲基转移酶缺乏症[guanidinoacetate methyltransferase(GAMT)deficiency]及肌酸转运子缺乏症[creatine transporter(CT)deficiency]。该组疾病的临床主要特点是智力低下与癫痫,提示大脑灰质主要受累,伴随大脑肌酸缺乏的生化异常(可由MRS证实)。
肌酸是含氮有机酸,主要在肾脏和肝脏经AGAT及GAMT的作用而生成,通过主动跨膜的肌酸转运系统(CT)而抵达需能高的组织中,如储存在骨骼肌和大脑。它的磷酸化形式(肌酸-磷酸或磷酸肌酸)参与ATP的合成,在肌酸/磷酸肌酸的细胞库内被利用,与肌酸激酶(creatine kinase,CK)及ATP/ADP共同提供高能磷酸缓冲系统。细胞内肌酸与肌酸磷酸盐是经非酶作用转化为肌酐,每日恒定更新体内1.5%的肌酸。肌酐从尿排出,每日尿排出的肌酐与体内总肌酸直接成比例。有三种先天代谢缺陷可导致肌酸缺乏。两种常染色体隐性遗传病影响肌酸的生物合成,即精氨酸:甘氨酸转脒酶缺乏症和胍基乙酸甲基转移酶缺乏症。第三种为X连锁肌酸转运子缺乏症,将肌酸转运至大脑和肌肉发生缺陷。
1.AGAT缺乏症(MIM#612718)
本病是由于L-精氨酸:甘氨酸转脒酶(AGAT)缺乏所致,该酶在肌酸生物合成中催化第一个限速反应,该反应是从精氨酸到甘氨酸产生胍基乙酸及鸟氨酸的胍基的可逆性转脒反应。缺陷导致胍基乙酸(GAA)在尿和血浆水平下降。本病为常染色体隐性遗传,GATM基因位于染色体15q15。
2.GAMT缺乏症(MIM#612736)
1994年由stÖckler等首先报道,为常染色隐性遗传病,是由于S-腺苷基-L-甲硫氨酸:N-胍基乙酸甲基转移酶(在肝、肾、胰腺表达,脑内及其他组织表达较少)缺乏所致。由此产生肌酸缺乏及胍基乙酸在尿、血浆和脑脊液中水平升高。
3.X连锁肌酸转运子(CRTR)缺乏症(MIM#300352)
本病是X连锁遗传病,由于CT缺乏所致。SLC6A8基因位于Xq 28,在人体多种组织中表达,在骨骼肌与肾脏中表达最高,在结肠、脑、心、睾丸及前列腺中表达较少。肌酸主要在肝脏合成,通过血液转运,再进入需要肌酸的组织,这需要克服巨大浓度梯度,由Na+及Cl-依赖的肌酸转运子完成。近年已发现一些家系有数个男性同胞儿患本病。大约三分之一患者为新发突变,缺陷导致尿肌酸/肌酐比值升高,GAAS水平正常。在智力障碍、癫痫发作、孤独症谱系障碍男性患者中约占1%~3.5%。
1.AGAT缺乏症(MIM#612718)
血中肌酐及氨基酸正常,胍基乙酸减少。尿中胍基乙酸显著降低。AGAT活性显著降低(成纤维及淋巴母细胞放射化学测定)。MRS显示脑中Cr/PCr峰消失。可进行GATM基因突变分析。
2.GAMT缺乏症(MIM#612736)
(1)生化检查
血、尿及脑脊液中肌酸(Cr)与肌酐(Crn)均减少。Cr的前体胍基乙酸(GAA)浓度在本病显著升高(血、CSF、尿)。
(2)影像学检查
MRS是诊断本病的可靠技术,质子MRS通过Cr/PCr共振峰消失而可诊断脑内Cr缺乏。31P-MRS还可通过出现平时没有的磷酸胍基乙酸峰确证脑内PCr的减低。虽然本病患者脑内完全没有Cr/PCr信号,但其肌肉中Cr的缺乏并不显著。
(3)基因检测
GAMT基因位于染色体19p13.3。
3.X连锁肌酸转运子(CRTR)缺乏症(MIM#300352)
血及尿中肌酸浓度升高,血肌酸酐正常,血及尿中胍基乙酸浓度正常。MRS证实脑内Cr/PCr信号消失。最近认为肯定诊断本病应进行成纤维细胞肌酸摄取实验,以区分患儿、携带者及正常人。还可进行CT基因突变分析。
1.AGAT缺乏症(MIM#612718)
口服补充单水肌酸300~400mg/(kg·d),3~9个月即可使MRS脑内Cr/PCr信号增加至正常的40%~80%,16个月后可达正常。治疗后可使患儿视觉感知及精细运动技巧快速进步,认知发育有缓慢进步,语言能力进步最慢。通过MRS分析大脑肌酸水平可用于监测治疗反应。
2.GAMT缺乏症(MIM#612736)
治疗有两个目的,降低GAA水平和补充大脑肌酸。大剂量单水肌酸400~800g/(kg·d),通过大剂量 L-鸟氨酸 400~800g/(kg·d)或减少底物限制精氨酸饮食,竞争性抑制AGAT活性,从而降低GAA水平。另外,苯甲酸钠可通过与甘氨酸结合形成马尿酸,由肾脏迅速排出,从而减少GAA的产生。
根据报道治疗反应包括锥体外系异常症候缓解、发育进步、癫痫好转等。治疗25个月后,脑内Cr可完全恢复,但患者在治疗后均不能达到完全发育正常。
治疗并不能完全纠正胍基乙酸贮积(具有神经毒性),所以饮食限制精氨酸(合成胍基乙酸的限速底物),并以鸟氨酸代替(完全抑制胍基乙酸合成)是两种辅助治疗措施。这两种措施必须同时进行,才可以永久的纠正胍基乙酸浓度的异常升高,并显著改善临床预后。
3.X连锁肌酸转运子(CRTR)缺乏症(MIM#300352)
治疗包括口服补充单水肌酸400mg/(kg·d),可充精氨酸400mg/(kg·d)和甘氨酸150mg/(kg·d),但是疗效有限。迄今尚无有效治疗方法。
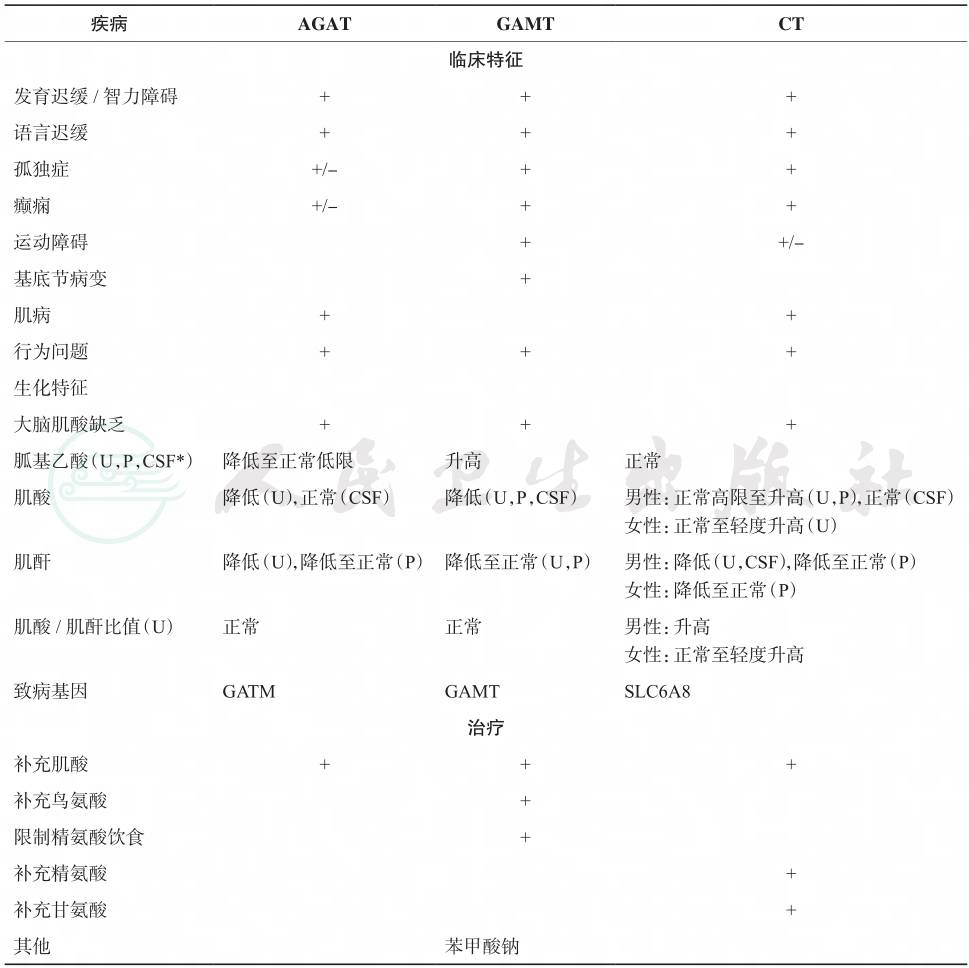
诊断:临床医生在遇到无法解释的智力低下、惊厥及语言发育延迟的患儿时,都要想到有无本病的可能性,对智力障碍伴孤独症样行为的患儿应该筛查肌酸缺乏综合征,各亚型间的比较见表1。
MRS检测肌酸峰和尿、血浆和/或脑脊液的GAA、肌酸、肌酐水平测定可作出诊断。检测体液中的胍基乙酸可以区别GAMT(高浓度)、AGAT(低浓度)及CT(正常浓度)缺乏症。体液中肌酸与肌酐比值的改变也是重要的生化特点。GAMT及AGAT缺乏症可以口服补充肌酸治疗,但CT缺乏症则对这种治疗疗效有限。
表1几种疾病的特征比较
引自:遗传代谢病防治理论与实践.第1版.ISBN:978-7-117-34392-3.主编:









