


 收藏
收藏 已收藏
已收藏英文名称 :dopa-responsive dystonia
中文别名 :多巴反应性肌张力不全
多巴反应性肌张力障碍(dopα-responsive dystonia,DRD),是一种具有明显昼夜波动性的遗传性进行性肌张力不全。该病由Segawa于1976年首先报道,于1988年由Nygaard等首先命名,国内病例于1997年首次报道。DRD的患病率为(0.5~1)/百万,其表型谱广泛,分为经典型和多巴反应性肌张力障碍附加症(DRD-plus)。经典型常见,多以足部肌张力不全为首发症状,可有轻度帕金森样表现,晨轻暮重,休息或睡眠后改善,小剂量左旋多巴治疗有效。现认为DRD是由基因突变引起的多巴胺合成途径异常不伴胶质细胞丢失的一类疾病。本病预后良好,延误治疗会严重影响预后。
DRD是由多巴胺合成不足导致的。参与多巴胺合成及循环的多个酶的缺陷,导致基底节突触末端多巴胺不足,易于耗竭,引起运动及非运动功能障碍。其致病基因包括GCH1、TH、PTS、SPR、PCBD、QDPR基因等。
GCH1和TH为本病的主要致病基因。
1.GCH1基因
是经典型DRD与DRD-plus最常见的致病基因,具有常染色体显性及常染色体隐性两种遗传方式。GCH1基因定位于14q22,编码三磷酸鸟苷环化水解酶1,是四氢生物蝶呤合成第一步的限速酶,而BH4不仅是酪氨酸羟化酶的辅酶,也是苯丙氨酸羟化酶的辅酶,因此,其缺乏不仅导致多巴胺缺乏,还可导致高苯丙氨酸血症。此外,BH4还是色氨酸羟化酶的辅酶,BH4的严重缺乏导致5-羟色胺神经元功能异常,而5-羟色胺神经元对于姿势的维持、运动的协调有重要作用,其功能异常导致肌张力低下,运动障碍。另外,5-羟色胺神经元通过调节突触形成影响皮层的发育与功能,可能是部分患者表现为DRD-plus及左旋多巴疗效有限的原因,合并应用5-羟色胺可使症状进一步改善。有研究依据遗传方式及临床表现将DRD划分为以下3类:①常染色体显性遗传/左旋多巴反应型:表现为典型DRD,儿童期起病,小剂量左旋多巴治疗效果显著;②常染色体隐性遗传伴高苯丙氨酸血症:表现为DRD-plus,起病早,伴有精神运动发育迟滞、惊厥等表现;③复合杂合突变(常染色体隐性遗传):其临床严重程度介于上述两种类型之间。
2.TH基因
定位于11p55,其突变导致的DRD呈常染色体隐性遗传。TH基因编码的酪氨酸羟化酶可以将酪氨酸转换为多巴胺,参与多巴胺合成。而多巴胺可进一步代谢成高香草酸及3-甲基-4-羟基苯氧乙醇,多巴胺合成减少进一步导致脑脊液中高香草酸及3-甲基-4-羟基苯氧乙醇浓度降低,而酪氨酸和5-羟吲哚乙酸浓度正常。另外,酪氨酸羟化酶还是儿茶酚胺类神经递质(去甲肾上腺素及肾上腺素)合成的限速酶,故该酶的缺陷可导致广泛性的神经功能异常,引起以下临床表现:①进展性婴幼儿脑病:表现为运动发育落后,波动性锥体外系症状,动眼危象及自主神经症状,左旋多巴可减轻上述症状,但不一定能完全缓解;②左旋多巴反应性婴幼儿帕金森,表现为婴幼儿期起病的严重的运动障碍,如帕金森样症状、非癫痫性肌阵挛发作及上睑下垂,左旋多巴对帕金森样症状效果显著,而2.5%的去氧肾上腺素眼部应用后可使上睑下垂明显好转;③经典型DRD:前两型归类于DRD-plus。由于酪氨酸羟化酶主要存在于大脑及肾上腺髓质,因此不能通过测定白细胞及成纤维细胞的酶活性进行诊断。目前主要通过基因突变分析确诊。
SPR基因遗传方式主要为常染色体隐性遗传,亦有常染色体显性遗传报道。其临床表现除典型DRD表现外还会引起发育迟缓,肌张力低下,动眼危象以及认知障碍。PTS基因临床表现分为3型,即严重型、轻型或外周型、暂时型。严重型在生后3个月后出现类似PKU的临床表现,还可伴有躯干肌张力低下、眼睑下垂、反应迟钝、运动障碍、嗜睡等症状;外周型仅表现为苯丙氨酸增高,无神经系统症状;暂时型者为6-丙酮酰四氢蝶呤合成酶成熟延迟所致,随着酶的完全成熟,临床表现逐渐消失。QDPR基因突变所致DRD为染色体隐性遗传。患者除具有与6-丙酮酰四氢生物蝶呤合成酶缺乏症相似的临床表现外,还伴有免疫功能低下,易反复感染。PCBD基因突变呈常染色体隐性遗传,临床即可表现为DRD,亦可有高苯丙氨酸血症等BH4缺乏相应的临床表现。
多巴反应性肌张力障碍的诊断主要辅助检查包括苯丙氨酸负荷试验与四氢生物蝶呤负荷试验、脑脊液神经递质检查、酶学分析,以及相关基因突变分析。
1.多巴胺试验性治疗
多年来,小剂量多巴胺治疗有效一直作为DRD诊断的首选方法。多巴胺的推荐剂量从1mg/(kg·d)起始,逐渐加量,大部分患儿症状完全缓解的最终剂量为4~5mg/(kg·d)。
2.苯丙氨酸/四氢生物蝶呤负荷试验
患者在食用低蛋白早餐2小时后,测定血浆中苯丙氨酸及酪氨酸浓度作为基线浓度,随后口服苯丙氨酸溶液(100mg/kg)后,分别留取1小时、2小时、4小时的血样,测定血浆氨基酸浓度。诊断多巴反应性肌张力障碍的标准为第4小时血样中,苯丙氨酸/酪氨酸>7.5,虽然苯丙氨酸负荷试验有助于多巴反应性肌张力障碍的诊断,但是难以区分是何种基因型所导致,也不能鉴别经典型PKU与DRD,后者可联合四氢生物蝶呤负荷试验进行鉴别,即在服用苯丙氨酸3小时后再口服四氢生物蝶呤20mg/kg,服后2、4、6、8、24小时采血测苯丙氨酸浓度。在血苯丙氨酸浓度 >600μmol/L情况下,可以直接进行四氢生物蝶呤负荷试验。四氢生物蝶呤缺乏者,当给予四氢生物蝶呤后,血苯丙氨酸明显下降:PTPS缺乏者,在服用四氢生物蝶呤后4~6小时血苯丙氨酸浓度下降至正常;DHPR缺乏者,血苯丙氨酸浓度一般在8小时或之后下降至正常。经典型PKU患者血苯丙氨酸浓度无明显变化。
3.影像学检查
多巴胺转运体(dopamine transporter,DAT)成像主要用于DRD与帕金森的鉴别。多巴胺转运体主要存在于多巴胺能神经元末端,多巴胺转运体成像可以检测纹状体多巴胺能神经元的完整性,帕金森患者存在胶质细胞及多巴胺能神经元缺失,而DRD患者无缺失,因此帕金森患者的多巴胺转运体成像往往减低,而DRD患者的多巴胺转运体成像正常。
4.脑脊液检查
多巴胺代谢通路中不同酶的缺乏,导致代谢产物在脑脊液中发生不同的改变,完善脑脊液生物蝶呤等代谢产物的测定,对于鉴别哪种基因突变所致的DRD有一定的指导意义(表1)。
表1不同基因突变所致的DRD脑脊液代谢产物改变
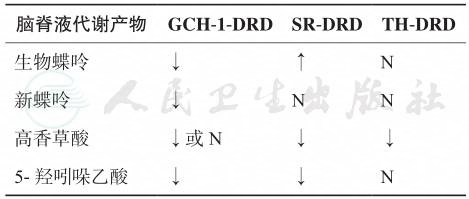
GCH-1-DRD:三磷酸鸟苷环化水解酶1缺乏导致的DRD;SR-DRD:墨蝶呤还原酶缺乏导致的DRD;TH-DRD:酪氨酸羟化酶缺乏导致的DRD;↑浓度升高;↓浓度降低
引自:遗传代谢病防治理论与实践.第1版.ISBN:978-7-117-34392-3.主编:
5.基因检测
基因检测是本病的确诊依据,尤其是在苯丙氨酸、四氢生物蝶呤负荷试验阴性或临界状态但临床高度怀疑DRD时。常见的DRD的致病基因包括GCH1、SPR及TH基因。明确致病基因对后续治疗亦有指导意义。
1.左旋多巴
小剂量的左旋多巴[1~5mg/(kg·d)]大多疗效显著且持久,尤其对肌张力不全效果明显,但对DRD-plus的非运动系统的症状(认知障碍等)效果欠佳。在儿童左旋多巴的推荐剂量为1mg/(kg·d)起始,逐渐加量,大部分患儿最终剂量控制在4~5mg/(kg·d)。成人一般从100mg/d开始缓慢加量,大部分患者有效剂量在300~400mg/d。最大剂量儿童为20mg/(kg·d),成人为1000mg/d。有文献报道,GCH1基因突变的患者有效平均剂量为166mg/d(25~400mg/d),非GCH1基因突变所需要的平均剂量为232mg/d(12.5~600mg/d)。推荐在服用左旋多巴的同时服用多巴胺脱羧酶抑制剂。
2.5-羟色胺
当左旋多巴单独治疗效果不理想时,可同时口服5-羟色胺1~8mg/(kg·d),分3~4次服用,从小剂量开始,缓慢加量,以数日或数周增加1mg/kg为宜。
3.其他
部分患儿需同时加用四氢生物蝶呤口服。此外,左旋多巴、5-羟色胺会导致脑脊液中叶酸水平降低,需要同时补充叶酸(15mg/d)。









