


 收藏
收藏 已收藏
已收藏英文名称 :congenital nephrogenic diabetes insipidus
英文名称 :congenital nephrogenic diabetes insipidus
先天性肾性尿崩症(congenital nephrogenic diabetes insipidus,CNDI)是一种罕见的遗传性疾病,约占肾性尿崩的10%左右,其发病率约为8.8/1 000 000。由于AVP受体(V2R)或AQP2 基因突变,使远端肾单位对精氨酸加压素(arginine vasopressin,AVP)的抗利尿作用出现抵抗,肾单位不能正常浓缩尿液,患者表现为多饮、多尿、烦渴,尿比重下降而继发引起脱水、高钠血症、生长发育迟缓等表现。本病在我国少有报道。
先天性肾性尿崩症(congenital nephrogenic diabetes insipidus,CNDI)是一种罕见的遗传性疾病,约占肾性尿崩的10%左右,其发病率约为8.8/1 000 000。由于AVP受体(V2R)或AQP2 基因突变,使远端肾单位对精氨酸加压素(arginine vasopressin,AVP)的抗利尿作用出现抵抗,肾单位不能正常浓缩尿液,患者表现为多饮、多尿、烦渴,尿比重下降而继发引起脱水、高钠血症、生长发育迟缓等表现。本病在我国少有报道。
肾脏是维持和调节体内水平衡的重要器官。这个调节过程主要在肾小管和集合管内发生,同时受AVP的调节。AVP是由垂体后叶分泌,通过血液循环到达远端肾单位,与肾脏远曲小管和集合管细胞基底外侧的AVP受体(V2R)结合。激活的V2R通过刺激Gs 蛋白和腺苷酸环化酶的磷酸化反应,催化ATP转化为cAMP,导致细胞内cAMP 水平升高,从而激活蛋白激酶A,使AQP2磷酸化,AQP2四聚体中的三个单体磷酸化使AQP2 同源四聚体重新分布,使AQP2从细胞质内的储存囊泡转运至顶膜,水分子从顶膜上的AQP3和AQP4进入血液循环,从而实现尿液浓缩。当AVP与V2R分离后,上述过程可发生逆转。由此可见,正常尿液的生成,除需要AVP之外,V2R和AQP2 的正常表达及发挥功能也至关重要。在CNDI中,AVP 水平正常或升高,容量感受器及渗透压感受器正常,cAMP产生减少,可能由于AVP受体(V2R)缺陷,不能与AVP结合。或者AQP2缺陷,因而对AVP无反应。从而导致先天性肾性尿崩的发生(图32-2)。
肾脏是维持和调节体内水平衡的重要器官。这个调节过程主要在肾小管和集合管内发生,同时受AVP的调节。AVP是由垂体后叶分泌,通过血液循环到达远端肾单位,与肾脏远曲小管和集合管细胞基底外侧的AVP受体(V2R)结合。激活的V2R通过刺激Gs 蛋白和腺苷酸环化酶的磷酸化反应,催化ATP转化为cAMP,导致细胞内cAMP 水平升高,从而激活蛋白激酶A,使AQP2磷酸化,AQP2四聚体中的三个单体磷酸化使AQP2 同源四聚体重新分布,使AQP2从细胞质内的储存囊泡转运至顶膜,水分子从顶膜上的AQP3和AQP4进入血液循环,从而实现尿液浓缩。当AVP与V2R分离后,上述过程可发生逆转。由此可见,正常尿液的生成,除需要AVP之外,V2R和AQP2 的正常表达及发挥功能也至关重要。在CNDI中,AVP 水平正常或升高,容量感受器及渗透压感受器正常,cAMP产生减少,可能由于AVP受体(V2R)缺陷,不能与AVP结合。或者AQP2缺陷,因而对AVP无反应。从而导致先天性肾性尿崩的发生(图32-2)。
研究表明,导致先天性肾性尿崩的病因为编码V2R或AQP2的基因突变引起肾脏尿液浓缩障碍。其中约有90%的遗传性肾性尿崩症是以X连锁方式隐性遗传,定位于染色体区域Xq28,另外10%的病例是由一个位于染色体12q13,编码AQP2基因发生突变引起的,以常染色体显性或常染色体隐性方式遗传,引起常染色体隐性遗传的AQP2基因突变约占其中的90%,不到10%为常染色体显性性遗传。
1992年,VR2基因克隆成功,并在一个肾性尿崩症患者发现此基因的突变。VR2基因序列长度大约2.2kp,其中包括3个外显子以及3’端非翻译序列。编码一个含有371个氨基酸的7跨膜区蛋白质,分子量为40518道尔顿。已报道的与VPR2相关的突变点至少有251种,其中至少21种突变不会导致疾病的发生。所有突变类型中,错义突变占48%,无义突变(> 13%),其次是小片段移码缺失(> 10%)。目前已经证实,AVPR2基因突变并非局限于受体蛋白基因的某一个区域,而是分散存在于整个基因片段的编码区。多数患者只要出现单个氨基酸的改变,就可以明显减少V2R受体在细胞膜上的数量或降低受体与AVP的亲和力。突变虽然不影响蛋白质的合成,但是显著降低了AVP受体与Gs蛋白的偶联,从而影响AVP对水的重吸收作用。
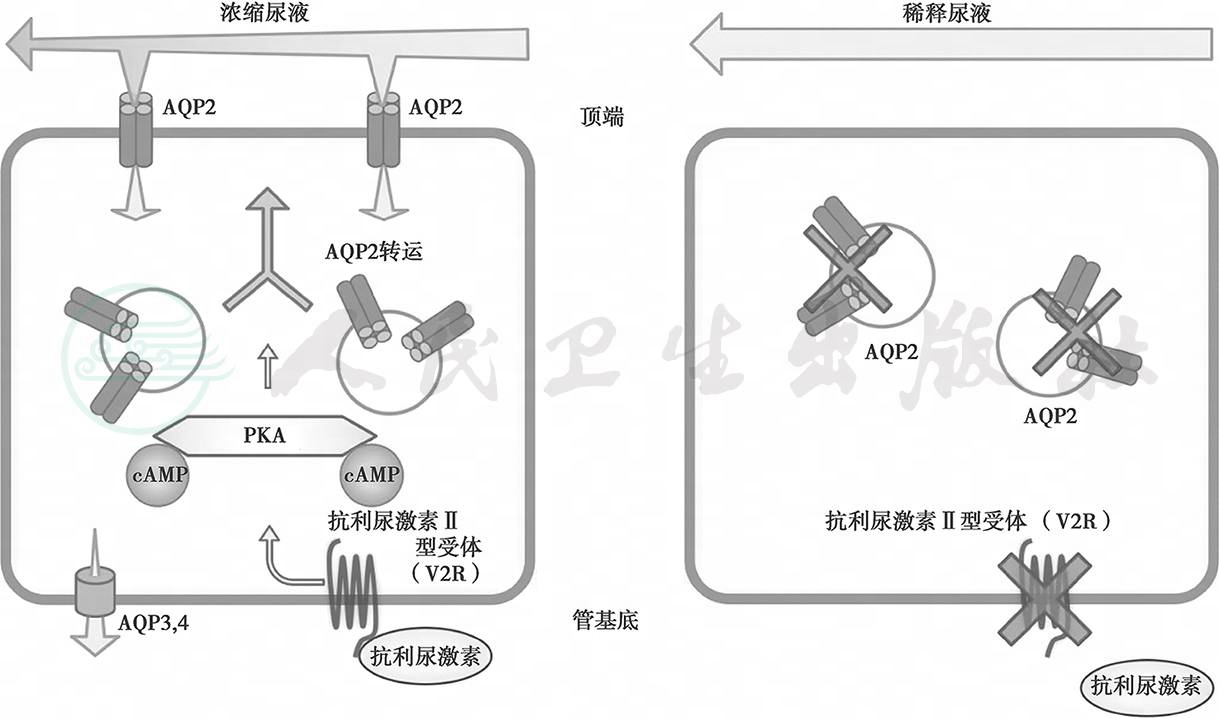
图32-2先天性肾性尿崩症发病机制
引自:遗传代谢病防治理论与实践.第1版.ISBN:978-7-117-34392-3.主编:
现已发现的AVPR2基因突变主要有4种不同的类型:①突变导致mRNA的合成受阻,使受体蛋白的合成减少;② 在蛋白质翻译过程中合成了异常蛋白质,使合成的蛋白质滞留在内质网中不能到达细胞膜表面;③ 突变影响了激素与受体的结合;④突变干扰了细胞膜上受体蛋白与Gs蛋白的偶联。体外实验显示大部分AVPR2基因突变导致编码的受体滞留在细胞内不能到达膜上,少数突变的受体能够到达细胞表面,但不能与AVP结合或是不能很好地引发胞内的cAMP信号通路。无论是V2R数量减少,还是其结构功能的改变,都使V2R不能介导正常AVP的作用,从而导致遗传性肾性尿崩症。
AQP2基因编码271个氨基酸残基组成的AQP2,分子量为28968 道尔顿。已报道的与AQP2相关的突变点约50个。AQP2基因突变,致使其在胞内的穿梭机制受损,使水通道功能缺陷,肾脏不能对AVP起反应而产生尿崩症。AQP2基因突变可引起常染色体隐性遗传性尿崩症和常染色体显性遗传性尿崩症,它们的发病机制不同。体外试验研究表明错误折叠使AQP2突变蛋白滞留在内质网,不能与野生型聚合。从而引起常染色体隐性遗传的先天性尿崩。而在显性遗传的肾性尿崩中AQP2突变蛋白离开内质网后能与野生型蛋白形成异源四聚体但其被高尔基体扣留,所以异源四聚体也就陷在这里了。常染色体显性遗传引起的突变蛋白不是没有功能,而是被高尔基体扣留阻碍足够的野生型蛋白到达集合管顶膜起作用。
研究表明,导致先天性肾性尿崩的病因为编码V2R或AQP2的基因突变引起肾脏尿液浓缩障碍。其中约有90%的遗传性肾性尿崩症是以X连锁方式隐性遗传,定位于染色体区域Xq28,另外10%的病例是由一个位于染色体12q13,编码AQP2基因发生突变引起的,以常染色体显性或常染色体隐性方式遗传,引起常染色体隐性遗传的AQP2基因突变约占其中的90%,不到10%为常染色体显性性遗传。
1992年,VR2基因克隆成功,并在一个肾性尿崩症患者发现此基因的突变。VR2基因序列长度大约2.2kp,其中包括3个外显子以及3’端非翻译序列。编码一个含有371个氨基酸的7跨膜区蛋白质,分子量为40518道尔顿。已报道的与VPR2相关的突变点至少有251种,其中至少21种突变不会导致疾病的发生。所有突变类型中,错义突变占48%,无义突变(> 13%),其次是小片段移码缺失(> 10%)。目前已经证实,AVPR2基因突变并非局限于受体蛋白基因的某一个区域,而是分散存在于整个基因片段的编码区。多数患者只要出现单个氨基酸的改变,就可以明显减少V2R受体在细胞膜上的数量或降低受体与AVP的亲和力。突变虽然不影响蛋白质的合成,但是显著降低了AVP受体与Gs蛋白的偶联,从而影响AVP对水的重吸收作用。
图32-2先天性肾性尿崩症发病机制
引自:遗传代谢病防治理论与实践.第1版.ISBN:978-7-117-34392-3.主编:
现已发现的AVPR2基因突变主要有4种不同的类型:①突变导致mRNA的合成受阻,使受体蛋白的合成减少;② 在蛋白质翻译过程中合成了异常蛋白质,使合成的蛋白质滞留在内质网中不能到达细胞膜表面;③ 突变影响了激素与受体的结合;④突变干扰了细胞膜上受体蛋白与Gs蛋白的偶联。体外实验显示大部分AVPR2基因突变导致编码的受体滞留在细胞内不能到达膜上,少数突变的受体能够到达细胞表面,但不能与AVP结合或是不能很好地引发胞内的cAMP信号通路。无论是V2R数量减少,还是其结构功能的改变,都使V2R不能介导正常AVP的作用,从而导致遗传性肾性尿崩症。
AQP2基因编码271个氨基酸残基组成的AQP2,分子量为28968 道尔顿。已报道的与AQP2相关的突变点约50个。AQP2基因突变,致使其在胞内的穿梭机制受损,使水通道功能缺陷,肾脏不能对AVP起反应而产生尿崩症。AQP2基因突变可引起常染色体隐性遗传性尿崩症和常染色体显性遗传性尿崩症,它们的发病机制不同。体外试验研究表明错误折叠使AQP2突变蛋白滞留在内质网,不能与野生型聚合。从而引起常染色体隐性遗传的先天性尿崩。而在显性遗传的肾性尿崩中AQP2突变蛋白离开内质网后能与野生型蛋白形成异源四聚体但其被高尔基体扣留,所以异源四聚体也就陷在这里了。常染色体显性遗传引起的突变蛋白不是没有功能,而是被高尔基体扣留阻碍足够的野生型蛋白到达集合管顶膜起作用。
(一)实验室检查
1.尿液检查
患儿尿量多,尿色清而无色,尿比重低,一般为1.001~1.005(约50~200mmol/L),尿糖、尿蛋白等为阴性。
2.电解质及血肾功能检查
患儿血钠常增高,部分患者血钠可正常,血浆渗透压多偏高或者正常。如肾脏受累,肾功能可有不同程度改变。
3.尿崩症症特殊试验检查
(1)禁水试验:
主要用于鉴别尿崩症和精神烦渴。方法:于早8时开始,试验前先排尿,测体重、尿量、尿比重及尿渗透压,测血钠和血浆渗透压。于1小时内给饮水20ml/kg,随后禁6~8小时,每小时收集尿液,测尿量、尿比重及尿渗透压。共收集6次,试验结束时采血测血钠及血浆渗透压。本试验过程必须密切严加观察,如果患者排尿甚多,虽禁饮还不到6小时,而体重已较原来下降5%,或者血压明显下降,立即停止试验。
正常人禁水后不出现严重的脱水症状,血浆渗透压变化不大,尿量明显减少,尿比重超过1.015,尿渗透压超过800mOsm/L,尿渗透压与血浆渗透压比率大于2.5;完全性尿崩症患者尿量无明显减少,比重小于1.010,尿渗透压小于280mOsm/L,血浆渗透压大于300mOsm/L,尿渗透压达300mOsm/L,或尿渗透压与血浆渗透压比率大于等于2,提示ADH分泌量正常,为精神性烦渴。
(2)禁水-加压试验:
用于中枢性尿崩与肾性尿崩症的鉴别。方法:先让患者禁水,每小时收集尿量一次,测其尿比重及渗透压情况。待连续两次尿渗透压差小于30mOsml/L时,注射水溶性加压素0.1U/kg,注射后每1时继续检测尿比重及渗透压,连续2~4次。正常人注射加压素后,尿渗透压可进一步升高;如用加压素后反应不良,尿量及比重、尿渗透压无明显变化可诊断肾性尿崩。
(3)血浆AVP定量:
用于与中枢性尿崩症相鉴别;中枢性尿崩患者血浆AVP低于正常;而肾性尿崩患者血浆AVP浓度增高或正常,但尿液不能浓缩而持续排出低渗尿,精神性烦渴症AVP分泌功能正常。但对病程久,病情重者可由于长期低渗状态,而使得AVP分泌障碍。
(4)基因检测:
对于怀疑肾性尿崩患者可进行AQp2基因和AVPR2基因突变的筛查确诊。
(二)影像学检查
头颅MRI检查:了解下丘脑及垂体的形态改变,排除颅脑肿瘤,中枢性尿崩患者垂体后叶高信号区消失,同时伴有侏儒症患者可发现垂体容量减少。
(一)实验室检查
1.尿液检查
患儿尿量多,尿色清而无色,尿比重低,一般为1.001~1.005(约50~200mmol/L),尿糖、尿蛋白等为阴性。
2.电解质及血肾功能检查
患儿血钠常增高,部分患者血钠可正常,血浆渗透压多偏高或者正常。如肾脏受累,肾功能可有不同程度改变。
3.尿崩症症特殊试验检查
(1)禁水试验:
主要用于鉴别尿崩症和精神烦渴。方法:于早8时开始,试验前先排尿,测体重、尿量、尿比重及尿渗透压,测血钠和血浆渗透压。于1小时内给饮水20ml/kg,随后禁6~8小时,每小时收集尿液,测尿量、尿比重及尿渗透压。共收集6次,试验结束时采血测血钠及血浆渗透压。本试验过程必须密切严加观察,如果患者排尿甚多,虽禁饮还不到6小时,而体重已较原来下降5%,或者血压明显下降,立即停止试验。
正常人禁水后不出现严重的脱水症状,血浆渗透压变化不大,尿量明显减少,尿比重超过1.015,尿渗透压超过800mOsm/L,尿渗透压与血浆渗透压比率大于2.5;完全性尿崩症患者尿量无明显减少,比重小于1.010,尿渗透压小于280mOsm/L,血浆渗透压大于300mOsm/L,尿渗透压达300mOsm/L,或尿渗透压与血浆渗透压比率大于等于2,提示ADH分泌量正常,为精神性烦渴。
(2)禁水-加压试验:
用于中枢性尿崩与肾性尿崩症的鉴别。方法:先让患者禁水,每小时收集尿量一次,测其尿比重及渗透压情况。待连续两次尿渗透压差小于30mOsml/L时,注射水溶性加压素0.1U/kg,注射后每1时继续检测尿比重及渗透压,连续2~4次。正常人注射加压素后,尿渗透压可进一步升高;如用加压素后反应不良,尿量及比重、尿渗透压无明显变化可诊断肾性尿崩。
(3)血浆AVP定量:
用于与中枢性尿崩症相鉴别;中枢性尿崩患者血浆AVP低于正常;而肾性尿崩患者血浆AVP浓度增高或正常,但尿液不能浓缩而持续排出低渗尿,精神性烦渴症AVP分泌功能正常。但对病程久,病情重者可由于长期低渗状态,而使得AVP分泌障碍。
(4)基因检测:
对于怀疑肾性尿崩患者可进行AQp2基因和AVPR2基因突变的筛查确诊。
(二)影像学检查
头颅MRI检查:了解下丘脑及垂体的形态改变,排除颅脑肿瘤,中枢性尿崩患者垂体后叶高信号区消失,同时伴有侏儒症患者可发现垂体容量减少。
目前对于遗传性肾性尿崩症无特效治疗药物,强调个体化的综合治疗。主要包括生活方式管理,维持和保证液体及水电解质的平衡,能量的供应,以及与传统药物相结合的综合治疗方案。包括合理的低盐饮食,合理饮水,定时排尿和传统药物治疗,旨在缓解症状,保证正常生长发育,预防并发症,减少药物不良反应。
1.低盐、合理蛋白饮食,合理饮水,定时排尿
先天性肾性尿崩症治疗方法主要为保证液体摄入量和适当限制钠盐及合理蛋白摄入。使容量和血钠在正常范围,并应注意提供足够的营养和热量,保证患儿生长发育正常和避免严重的脱水。早期治疗可减轻对生长和智能发育的影响。年长儿钠盐摄入应在 2~2.5mmol/(kg·d)。为了保证患儿生长发育的需求,合理蛋白质摄入的同时应适当提高食物中的碳水化合物及脂肪的比值,蛋白质推荐摄入量约2g/(kg·d),同时适当限制磷的摄入量。对于年长儿,每天保证300~400ml/(kg·d)水的摄入。由于婴儿和非常年幼的儿童不能独立地对口渴感增强做出反应,故应以每2小时1次的频率全天候地为其提供水分。严重病例可能需要持续胃饲。
2.传统药物治疗
主要包括:利尿剂(噻嗪类、保钾利尿剂)、前列腺素合成抑制剂及钠通道阻断剂等。
(1)利尿剂:
可导致钠的负平衡,使尿量减少和尿浓度上升。其作用机制尚不明了。噻嗪类利尿剂常用的药物主要有氢氯噻嗪2~3mg/(kg·d);推测机制可能为其抑制碳酸酐酶活性,减少近端小管对Na+ 的重吸收,通过管球反馈机制减少肾血流量,减少到达集合管的原尿量。保钾类利尿剂的作用机制可能为在远端小管和集合管抑制钠通道ENac的转运提高肾小管的的渗透压,进而提高尿中的渗透压而起效。运用此类药物注意维持血钾在正常水平。
(2)前列腺素合成抑制剂:
如吲哚美辛2mg/(kg·d)等可抑制肾性前列腺素的产生而减少多尿,增加尿比重的作用,尤其在起始应用时效果明显,机制可能与促进AVP 刺激的cAMP 生成和增加非AVP 依赖的溶质重吸收相关。且因其对肾功能有不良影响,故应慎用,常用于其他疗法无效者。
(3)钠离子通道阻断剂:
阿米洛利0.3mg/(kg·d),分3次口服,可以阻断连接管和集合管起始段的阿米洛利敏感性钠离子通道,抑制Na+的重吸收,减少血容量从而达到控制尿量的效果,且对于健康个体没有抗利尿作用。
氢氯噻嗪联合阿米洛利可以在增加疗效的基础上,减少低钾血症的发生,是目前肾性尿崩的临床一线用药,氢氯噻嗪联合吲哚美辛也可以达到相似的效果,注意胃肠道及肾损害等不良反应的发生。开始药物治疗后应该注意避免大量饮水造成水中毒的发生。定期评估多饮多尿症状、血电解质、血尿渗透压、泌尿系超声及生长发育等情况,对于指导生活习惯和临床用药具有重要意义。
3.其他
近年来针对肾性尿崩的研究为遗传性NDI的治疗提供了一些新的策略,但这些药物研究目前仅停留在体外试验或体内外实验阶段。主要有用于恢复AVPR2 功能的药物:
(1)分子伴侣(包括内质网滞留蛋白或细胞质折叠蛋白):
如巯基氧化还原酶和热休克蛋白。
(2)化学伴侣:
能帮助蛋白质折叠的非肽类分子,包括一些渗透物如甘油、二甲基亚砜和三甲胺N-氧化物等,但其毒性大、特异性低而限制了其在人体的应用。
(3)药物伴侣:
也是非肽类,细胞可渗透性配体,能协助蛋白质的折叠,因可直接结合内质网内特异性靶蛋白而不同于化学伴侣,能稳定其构象并促进向质膜的修复,同时降低了副反应的发生。结合突变受体的药物伴侣能否被激动剂置换是其功能修复能否成功的关键因素。在肾脏,AVP 与AVPR2 作用诱导水的重吸收旁路途径增加细胞膜上AQP2 的表达,目前尚无特异性药物。选择性cGMP PDE5抑制剂枸橼酸西地那非可阻止cGMP降解,使AQP2在细胞膜表达增加。最近有实验表明,枸橼酸西地那非可缓解锂诱导的CNDI的多尿症状。降钙素通过七个跨膜受体起作用,七个跨膜受体与GaS结合可增加细胞内的AQP2水平。体内和体外实验已证实降钙素通过cAMP调节机制诱导AVPR2在胞膜积聚。前列腺素 E2(PGE2)和类前列腺素 E2 受体(EP2)可通过不同途径增加AQP2 磷酸化和向顶膜的转运作用;此外,EP2 选择性激动剂可以部分弥补非功能性AVPR2的作用,为HNDI 的治疗开拓了新方向。主要是依靠细胞膜上AQP2 的聚磷酸化而实现。功能性AVPR2 缺失所致水重吸收障碍,归根结底是AVP 依赖性的AQP2 表达减少和向顶膜转运的缺失。因此,治疗这类疾病还可以依靠AVPR2 以外的旁路途径来增加细胞膜上AQP2的表达,目前尚无特异性药物。近期研究表明,二甲双胍可能是治疗NDI 的又一新型药。体外实验证实二甲双胍通过激活AMPK 调节AQP2 和尿素转运体A1(UT-A1)的磷酸化,激活蛋白转运功能,改善MDCK 细胞的尿液浓缩能力,体内小鼠实验进一步验证了这一机制。目前二甲双胍治疗NDI 的临床药物试验正在进行。近期体内外研究发现他汀类可以促进AQP2 在细胞顶端膜的表达,潜在的分子机制尚未完全阐明,可能与Rho家族蛋白的异戊酰化调节AQP2 转运、调控细胞骨架相关。他汀类对NDI 患者的治疗效果目前尚不明确,可能提供新的治疗策略。与此同时,针对遗传性肾性尿崩症的基因治疗也在如火如荼的研究当中。基因治疗可能是彻底治愈先天性NDI的潜在手段,CRISPR-Cas9 基因编辑系统强大的基因编辑能力使遗传性疾病的治愈成为可能。
4.随访
先天性肾性尿崩症属于终生需要治疗疾病,故需要定期随访,以便观察治疗效果,及时调整治疗方案,预防并发症。婴幼儿期每1~2周随访一次,幼儿期后每月随访一次,每次均需查尿常规,血、尿比重,肝肾功能;每年至少复查一次泌尿系统彩超,动态评估患儿生长发育及并发症情况。
目前对于遗传性肾性尿崩症无特效治疗药物,强调个体化的综合治疗。主要包括生活方式管理,维持和保证液体及水电解质的平衡,能量的供应,以及与传统药物相结合的综合治疗方案。包括合理的低盐饮食,合理饮水,定时排尿和传统药物治疗,旨在缓解症状,保证正常生长发育,预防并发症,减少药物不良反应。
1.低盐、合理蛋白饮食,合理饮水,定时排尿
先天性肾性尿崩症治疗方法主要为保证液体摄入量和适当限制钠盐及合理蛋白摄入。使容量和血钠在正常范围,并应注意提供足够的营养和热量,保证患儿生长发育正常和避免严重的脱水。早期治疗可减轻对生长和智能发育的影响。年长儿钠盐摄入应在 2~2.5mmol/(kg·d)。为了保证患儿生长发育的需求,合理蛋白质摄入的同时应适当提高食物中的碳水化合物及脂肪的比值,蛋白质推荐摄入量约2g/(kg·d),同时适当限制磷的摄入量。对于年长儿,每天保证300~400ml/(kg·d)水的摄入。由于婴儿和非常年幼的儿童不能独立地对口渴感增强做出反应,故应以每2小时1次的频率全天候地为其提供水分。严重病例可能需要持续胃饲。
2.传统药物治疗
主要包括:利尿剂(噻嗪类、保钾利尿剂)、前列腺素合成抑制剂及钠通道阻断剂等。
(1)利尿剂:
可导致钠的负平衡,使尿量减少和尿浓度上升。其作用机制尚不明了。噻嗪类利尿剂常用的药物主要有氢氯噻嗪2~3mg/(kg·d);推测机制可能为其抑制碳酸酐酶活性,减少近端小管对Na+ 的重吸收,通过管球反馈机制减少肾血流量,减少到达集合管的原尿量。保钾类利尿剂的作用机制可能为在远端小管和集合管抑制钠通道ENac的转运提高肾小管的的渗透压,进而提高尿中的渗透压而起效。运用此类药物注意维持血钾在正常水平。
(2)前列腺素合成抑制剂:
如吲哚美辛2mg/(kg·d)等可抑制肾性前列腺素的产生而减少多尿,增加尿比重的作用,尤其在起始应用时效果明显,机制可能与促进AVP 刺激的cAMP 生成和增加非AVP 依赖的溶质重吸收相关。且因其对肾功能有不良影响,故应慎用,常用于其他疗法无效者。
(3)钠离子通道阻断剂:
阿米洛利0.3mg/(kg·d),分3次口服,可以阻断连接管和集合管起始段的阿米洛利敏感性钠离子通道,抑制Na+的重吸收,减少血容量从而达到控制尿量的效果,且对于健康个体没有抗利尿作用。
氢氯噻嗪联合阿米洛利可以在增加疗效的基础上,减少低钾血症的发生,是目前肾性尿崩的临床一线用药,氢氯噻嗪联合吲哚美辛也可以达到相似的效果,注意胃肠道及肾损害等不良反应的发生。开始药物治疗后应该注意避免大量饮水造成水中毒的发生。定期评估多饮多尿症状、血电解质、血尿渗透压、泌尿系超声及生长发育等情况,对于指导生活习惯和临床用药具有重要意义。
3.其他
近年来针对肾性尿崩的研究为遗传性NDI的治疗提供了一些新的策略,但这些药物研究目前仅停留在体外试验或体内外实验阶段。主要有用于恢复AVPR2 功能的药物:
(1)分子伴侣(包括内质网滞留蛋白或细胞质折叠蛋白):
如巯基氧化还原酶和热休克蛋白。
(2)化学伴侣:
能帮助蛋白质折叠的非肽类分子,包括一些渗透物如甘油、二甲基亚砜和三甲胺N-氧化物等,但其毒性大、特异性低而限制了其在人体的应用。
(3)药物伴侣:
也是非肽类,细胞可渗透性配体,能协助蛋白质的折叠,因可直接结合内质网内特异性靶蛋白而不同于化学伴侣,能稳定其构象并促进向质膜的修复,同时降低了副反应的发生。结合突变受体的药物伴侣能否被激动剂置换是其功能修复能否成功的关键因素。在肾脏,AVP 与AVPR2 作用诱导水的重吸收旁路途径增加细胞膜上AQP2 的表达,目前尚无特异性药物。选择性cGMP PDE5抑制剂枸橼酸西地那非可阻止cGMP降解,使AQP2在细胞膜表达增加。最近有实验表明,枸橼酸西地那非可缓解锂诱导的CNDI的多尿症状。降钙素通过七个跨膜受体起作用,七个跨膜受体与GaS结合可增加细胞内的AQP2水平。体内和体外实验已证实降钙素通过cAMP调节机制诱导AVPR2在胞膜积聚。前列腺素 E2(PGE2)和类前列腺素 E2 受体(EP2)可通过不同途径增加AQP2 磷酸化和向顶膜的转运作用;此外,EP2 选择性激动剂可以部分弥补非功能性AVPR2的作用,为HNDI 的治疗开拓了新方向。主要是依靠细胞膜上AQP2 的聚磷酸化而实现。功能性AVPR2 缺失所致水重吸收障碍,归根结底是AVP 依赖性的AQP2 表达减少和向顶膜转运的缺失。因此,治疗这类疾病还可以依靠AVPR2 以外的旁路途径来增加细胞膜上AQP2的表达,目前尚无特异性药物。近期研究表明,二甲双胍可能是治疗NDI 的又一新型药。体外实验证实二甲双胍通过激活AMPK 调节AQP2 和尿素转运体A1(UT-A1)的磷酸化,激活蛋白转运功能,改善MDCK 细胞的尿液浓缩能力,体内小鼠实验进一步验证了这一机制。目前二甲双胍治疗NDI 的临床药物试验正在进行。近期体内外研究发现他汀类可以促进AQP2 在细胞顶端膜的表达,潜在的分子机制尚未完全阐明,可能与Rho家族蛋白的异戊酰化调节AQP2 转运、调控细胞骨架相关。他汀类对NDI 患者的治疗效果目前尚不明确,可能提供新的治疗策略。与此同时,针对遗传性肾性尿崩症的基因治疗也在如火如荼的研究当中。基因治疗可能是彻底治愈先天性NDI的潜在手段,CRISPR-Cas9 基因编辑系统强大的基因编辑能力使遗传性疾病的治愈成为可能。
4.随访
先天性肾性尿崩症属于终生需要治疗疾病,故需要定期随访,以便观察治疗效果,及时调整治疗方案,预防并发症。婴幼儿期每1~2周随访一次,幼儿期后每月随访一次,每次均需查尿常规,血、尿比重,肝肾功能;每年至少复查一次泌尿系统彩超,动态评估患儿生长发育及并发症情况。
NDI通常以X连锁方式遗传(约90%的个体),也可以以常染色体隐性方式遗传(约9%的个体)或以常染色体显性方式遗传(约1%的个体)。同胞和后代的风险取决于父母的遗传方式及突变基因携带情况,目前可以通过分子诊断确定。如果家族中有致病突变基因被鉴定,产前检测可用于高危妊娠人群。
1.先天性肾性尿崩症患者如为X连锁隐性遗传,若患儿母亲为突变基因携带者,其生产的男孩50%可出现本病症,女儿为基因携带者,虽无临床症状,但尿浓缩功能可能出现异常,基因携带者所生的男性有50%可能发病。对于常染色体隐性遗传者,若夫妻双方均为携带者者其子女患病率约为25%,常染色体隐性遗传,父母一方为携带者,子代有50%为携带者;若为常染色体显性遗传患者,其子女患病概率与夫妻基因型有关;若夫妻均为杂合子,其子代患病率为75%;若夫妻均为纯合子或其一方为杂合子,其子代患病率为100%。
2.优生优育措施
(1)避免近亲结婚。
(2)产前诊断:对有先天性肾性尿崩高危家庭中高危人群,可在妊娠16~20孕周时经羊水穿刺或10~12孕周经绒毛膜绒毛取样提取胎儿细胞的DNA,可对突变已知家系进行基因产前诊断。对有本病家族史的夫妇及先证者可进行 DNA 分析,并对其胎儿进行产前诊断。家族成员基因分析也可检出杂合子携带者,进行遗传咨询。
(3)开展新生儿筛查及早发现先天性肾性尿崩患儿,尽早开始治疗,减少并发症及不良预后。
3.协调长期管理患者 向家长介绍有关先天性肾性尿崩症的相关知识、治疗方案、预后、预防、生活管理等。
NDI通常以X连锁方式遗传(约90%的个体),也可以以常染色体隐性方式遗传(约9%的个体)或以常染色体显性方式遗传(约1%的个体)。同胞和后代的风险取决于父母的遗传方式及突变基因携带情况,目前可以通过分子诊断确定。如果家族中有致病突变基因被鉴定,产前检测可用于高危妊娠人群。
1.先天性肾性尿崩症患者如为X连锁隐性遗传,若患儿母亲为突变基因携带者,其生产的男孩50%可出现本病症,女儿为基因携带者,虽无临床症状,但尿浓缩功能可能出现异常,基因携带者所生的男性有50%可能发病。对于常染色体隐性遗传者,若夫妻双方均为携带者者其子女患病率约为25%,常染色体隐性遗传,父母一方为携带者,子代有50%为携带者;若为常染色体显性遗传患者,其子女患病概率与夫妻基因型有关;若夫妻均为杂合子,其子代患病率为75%;若夫妻均为纯合子或其一方为杂合子,其子代患病率为100%。
2.优生优育措施
(1)避免近亲结婚。
(2)产前诊断:对有先天性肾性尿崩高危家庭中高危人群,可在妊娠16~20孕周时经羊水穿刺或10~12孕周经绒毛膜绒毛取样提取胎儿细胞的DNA,可对突变已知家系进行基因产前诊断。对有本病家族史的夫妇及先证者可进行 DNA 分析,并对其胎儿进行产前诊断。家族成员基因分析也可检出杂合子携带者,进行遗传咨询。
(3)开展新生儿筛查及早发现先天性肾性尿崩患儿,尽早开始治疗,减少并发症及不良预后。
3.协调长期管理患者 向家长介绍有关先天性肾性尿崩症的相关知识、治疗方案、预后、预防、生活管理等。









