


 收藏
收藏 已收藏
已收藏巨输尿管症由原发或继发的病因引起输尿管严重扩张积水。原发性巨输尿管症可分为梗阻性、反流性和非梗阻非反流性三类。先天性巨输尿管症指原发性梗阻性巨输尿管而言。反流性巨输尿管是由于膀胱输尿管反流,导致输尿管扩张积水。非梗阻非反流性巨输尿管是由于梗阻和反流以外的因素所致,可见于某些梨状腹综合征患者,偶见于尿崩症和尿路感染者。继发性巨输尿管症多源于膀胱内高压,如尿道瓣膜、神经源性膀胱、膀胱肿瘤等。
先天性巨输尿管症的病因及发病机制至今尚无统一认识,部分学者认为其主要病因为:胚胎期肾脏上升速度不及输尿管发育迅速,使得输尿管迂曲、引流不畅,导致输尿管外膜结缔组织增生,使输尿管蠕动波向下传递的过程中有所减弱,尿液对下端(病变部位)输尿管牵拉,引起逆向蠕动,导致功能性梗阻;若疾病持续进展,输尿管肌层(特别是纵行肌)出现压迫性萎缩伴慢性炎症、胶原纤维增生,最终使输尿管肾盂扩张积水。Mackinon等认为是末端输尿管内肌层结构异常,即环肌增厚和纵肌缺乏。Notley用电子显微镜检查异常输尿管肌层的神经分布是正常的,但肌层内有异常的胶原纤维,干扰了融合细胞层的排列,从而阻碍了蠕动的传导。由于CM的发生可能存在多种因素的参与,其发病机制与病理之间的关系尚无统一标准,然而已经通过组织学得到证实的因素包括:①输尿管下段平滑肌增生伴黏膜层及黏膜下层炎症发生;②输尿管下段环行肌正常而纵行肌缺失;③输尿管下段肌束/胶原纤维失调从而干扰正常融合细胞层样排列,蠕动波无法正常传递。
本病多见于男性,男∶女为3.5∶1~5∶1,左侧多见,左∶右为2∶1~3∶1,双侧占15%~25%。
先天性巨输尿管症多数是由于输尿管末端存在长0.5~4cm的无功能段,有组织学和超微结构上的异常,如管壁螺旋状肌层缺如、环肌增厚、胶原纤维明显增生,影响输尿管蠕动波的传导,导致功能性梗阻。输尿管开口往往看上去正常,输尿管导管大多可以顺利地通过,较少遇到真性狭窄。末端梗阻段以上输尿管扩张,下段更为明显,重症病例输尿管全程极度扩张,管径与小肠相仿,并伸长迂曲。肾积水及肾功能损害程度不等,易并发尿路感染。
巨输尿管症按病因分为4种类型:①反流性;②梗阻性;③反流伴梗阻性;④非反流非梗阻性。每种类型又可以分为原发性和继发性两种亚型。绝大多数先天性巨输尿管症属于原发性非反流非梗阻性。鉴别是否存在梗阻因素是决定治疗方案的重要依据,也是目前最大的争议。
目前普遍认为先天性巨输尿管症是由输尿管末端肌肉发育异常引起的。输尿管壁肌层于胎儿16~22周开始出现,由输尿管近端向远端发育。胎儿输尿管壁肌层主要由环形肌组成,出生后逐渐发育为环形、纵行双肌层。末端输尿管肌肉发育异常,包括环形肌肥厚、纵行肌缺乏、肌层胶原纤维增生等,导致该段输尿管的蠕动减弱或消失、尿液排泄不畅、输尿管管腔内压力升高,从而引起末端输尿管扩张,甚至向上传递引起近端输尿管扩张和肾积水。
磁共振成像及静脉尿路造影可见上述输尿管形态学改变,在排尿性膀胱尿道造影排除膀胱输尿管反流和下尿路病变后可明确诊断。胎儿超声可以早期诊断先天性巨输尿管症。受累患儿出生后应该接受一次完整的泌尿系统检查。
先天性巨输尿管症的治疗原则是去除病因、解除梗阻和保护肾功能。治疗方式的选择取决于发病年龄、上尿路积水程度和分肾功能,近十多年来保守治疗有增加趋势。Keating等认为87%的巨输尿管症患者可以选择保守治疗。DI RD等通过对保守治疗组患儿的长期随访,发现73%的患儿不需要手术干预。Simoni等认为手术适应证包括:①症状难以耐受;②肾功能恶化;③抗生素难以控制的泌尿道感染。
(一)输尿管膀胱再植术
手术方法为切除发育不全的输尿管末端,裁剪缩窄管腔使接近正常口径,松解游离上段输尿管,解除输尿管扭曲,将输尿管包埋于膀胱浆肌层下隧道里,将新的输尿管开口与三角区肌层边缘缝合。
Paquin认为膀胱壁内隧道长度超过输尿管直径5倍才能保证术后良好的抗反流效果。最常使用的输尿管裁剪方式是Starr的折叠法(图1)和Hendren的锥形裁剪法(图2)。然而 Ben-Meir等认为是否行输尿管裁剪对手术成功率没有明显的影响(裁剪组94%,非裁剪组96%)。与膀胱外输尿管膀胱再植术(如Lich-Gregoir法)相比,膀胱内输尿管膀胱再植术(如Cohen法、Politano-Leadbetter法(图3)手术效果更好,尤其适用于伴有膀胱功能受损的大龄儿童。Casale等的一项研究表明,保留输尿管周围神经的机器人辅助输尿管再植术能够获得更好的膀胱功能。
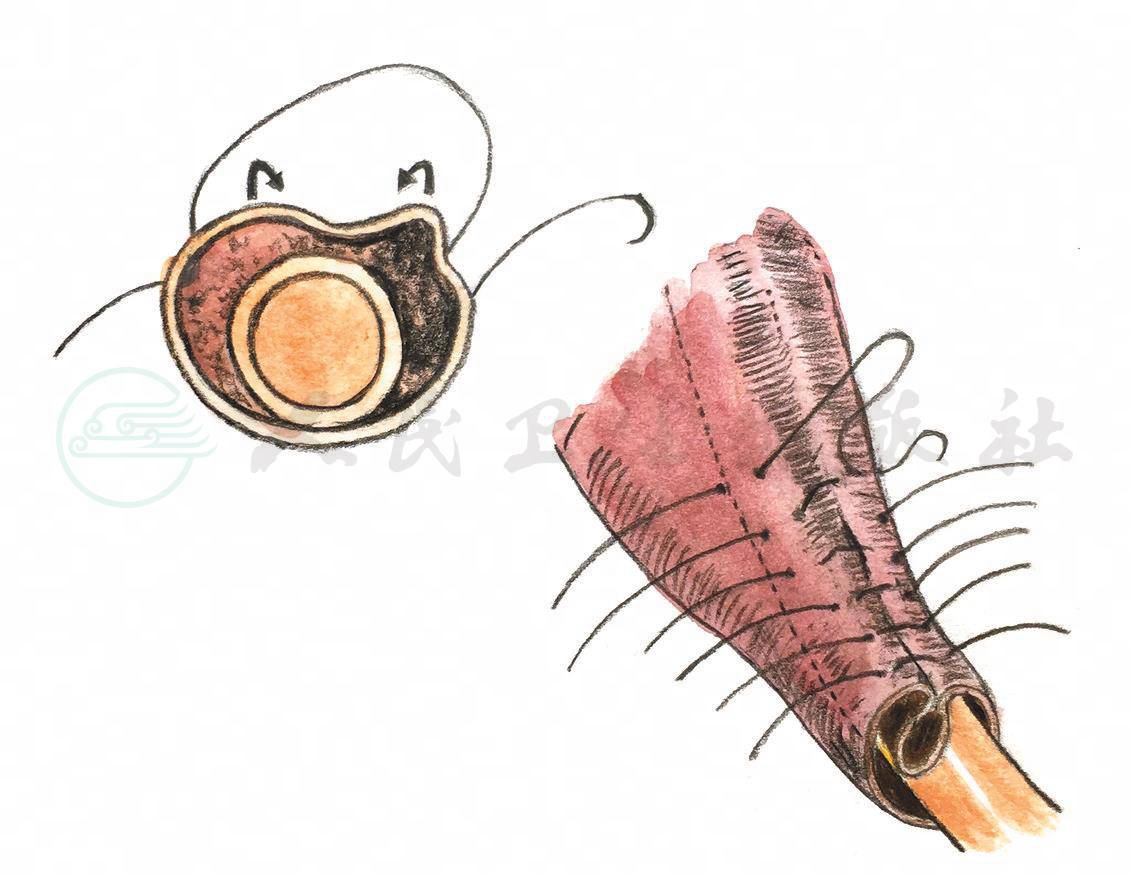
图1Starr折叠法5-0单股可吸收缝线间断内翻缝合
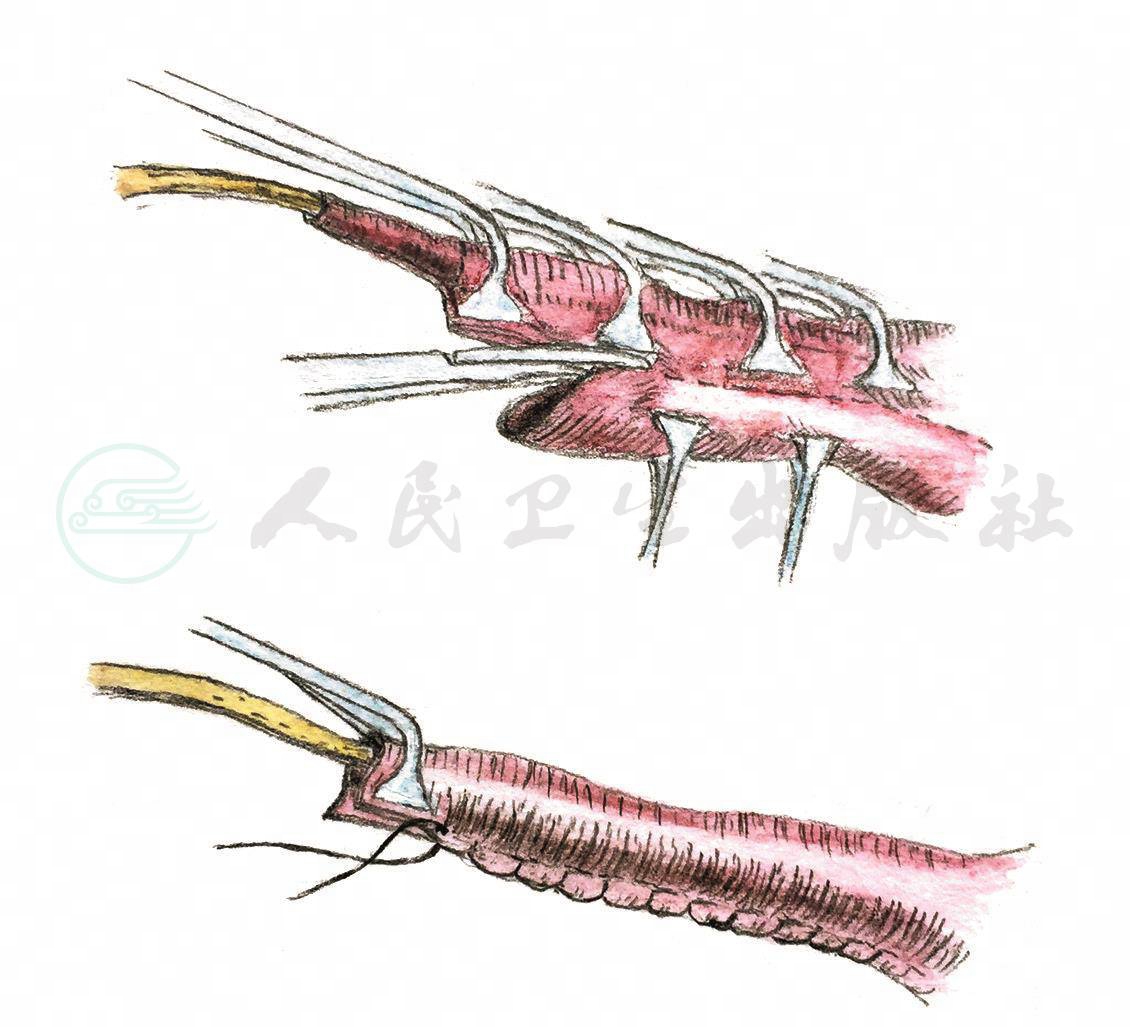
图2Hendren锥形裁剪法
置入输尿管导管(婴幼儿8-Fr,大龄儿童及成人10-Fr),沿无损伤钳边缘裁剪,5-0单股可吸收线缝合创面
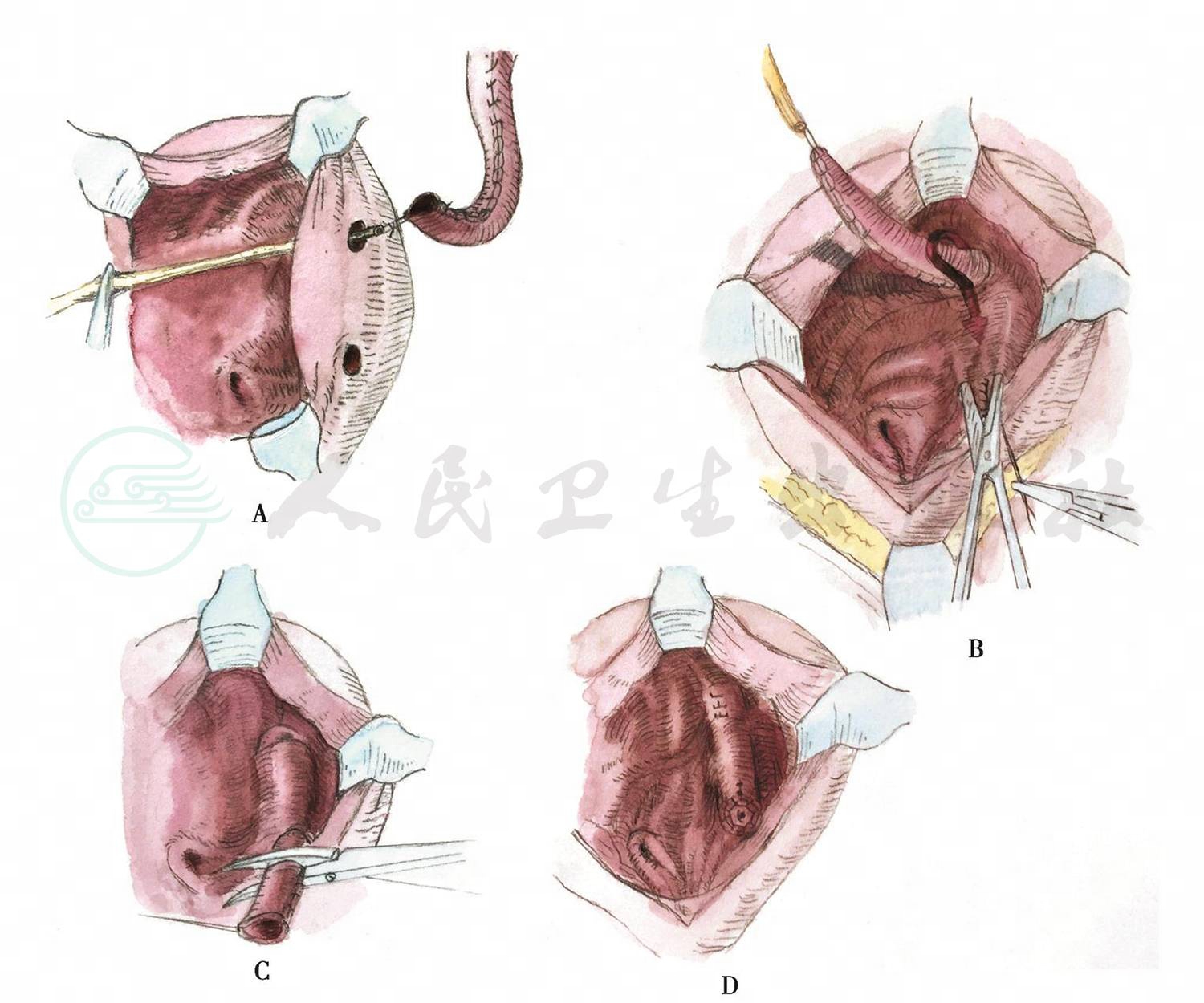
图3膀胱内输尿管膀胱再植术
A.输尿管拉入膀胱;B.穿过黏膜下隧道;C.剪除多余输尿管;D.输尿管口吻合固定
(二)肾穿刺造瘘和输尿管双J导管置入术
1999年Shenoy和Rance首先报道使用双J导管置入术治疗巨输尿管症,Farrugia等使用4.7F或5.2F双J导管治疗婴儿期巨输尿管症,半年换管一次,发现31%的患儿在置管期间出现并发症(双J导管移位、结石形成、感染),56%的患儿拔管后尿液引流功能恢复,不需要进一步的手术干预。
肾造瘘作为一种暂时性治疗方案适用于重度积水、全身状况差的患儿,能够迅速解除梗阻和改善肾积水。然而,肾造瘘术后有31%的患儿出现肾盂肾炎,双肾造瘘会引起膀胱功能减退甚至膀胱挛缩。因此,待患儿全身情况好转后,应该及时行输尿管再植术。
(三)肾脏、输尿管切除术
严重的巨输尿管症,肾功能损害严重且无法恢复,反复伴发感染的患儿,如果对侧肾脏功能良好,可行患侧肾脏、输尿管切除术。









