


 收藏
收藏 已收藏
已收藏英文名称 :homocystinuria
同型胱氨酸尿症(homocystinuria),又称假性马方综合征,是一种常染色体隐性遗传病,是甲硫氨酸代谢过程中由于酶缺乏,甲硫氨酸代谢紊乱而所致的疾病,是一种含硫氨基酸的先天性代谢障碍性疾病。同型胱氨酸尿症在世界范围内的发病率约为1/25万。
甲硫氨酸为一种必需氨基酸,约占饮食蛋白中氨基酸的2.5%,在体内一部分合成组织蛋白,其余部分主要转换途径是把分子中的硫转给L-同型半胱氨酸,再进一步转化为胱氨酸。甲硫氨酸转化为同型半胱氨酸的第一步是形成S-腺苷甲硫氨酸,该反应由甲硫氨酸腺苷转移酶催化。正常时,ATP分子的腺苷被转移到甲硫氨酸上,形成腺苷甲硫氨酸,这样就能参与几种转移甲基的反应。在正常人细胞内,S-腺苷甲硫氨酸转甲基形成S-腺苷同型半胱氨酸,后者可很快水解成同型半胱氨酸,并可与丝氨酸合成胱硫醚,其间经过一步转硫反应也可再甲基化而形成甲硫氨酸或氧化为同型胱氨酸(图1)。现已发现,在这种转化过程中,有多种酶的缺陷导致正常代谢途径中断,经异常代谢途径生成甲硫氨酸、同型胱氨酸,过多的同型胱氨酸激活凝血因子,抑制胶原的形成,引起结缔组织异常,病变可累及各个系统,以血管损害为主。其主要的临床表现是多发性血栓栓塞、智力落后、晶状体异位和指/趾过长,故有“假性马方综合征”之称。
同型胱氨酸尿症根据其酶缺陷不同可分为下列3型。
1.胱硫醚β合成酶(cystathione β-synthase,CBS)缺乏型(合成酶型)
此型最多见,包含维生素B6反应和无反应两种情况。由于从同型半胱氨酸转化为胱硫醚的代谢途径发生阻碍,因而血和尿中同型胱氨酸和甲硫氨酸浓度都升高。甲硫氨酸经中间代谢产物S-腺苷甲硫氨酸和S-腺苷同型半胱氨酸转化为同型半胱氨酸,后者可被氧化成同型胱氨酸或丝氨酸并结合成胱硫醚。形成胱硫醚的反应受胱硫醚β合成酶催化,该酶需维生素B6作为辅酶。因此,对维生素B6反应的患者使用大剂量维生素B6治疗有效。
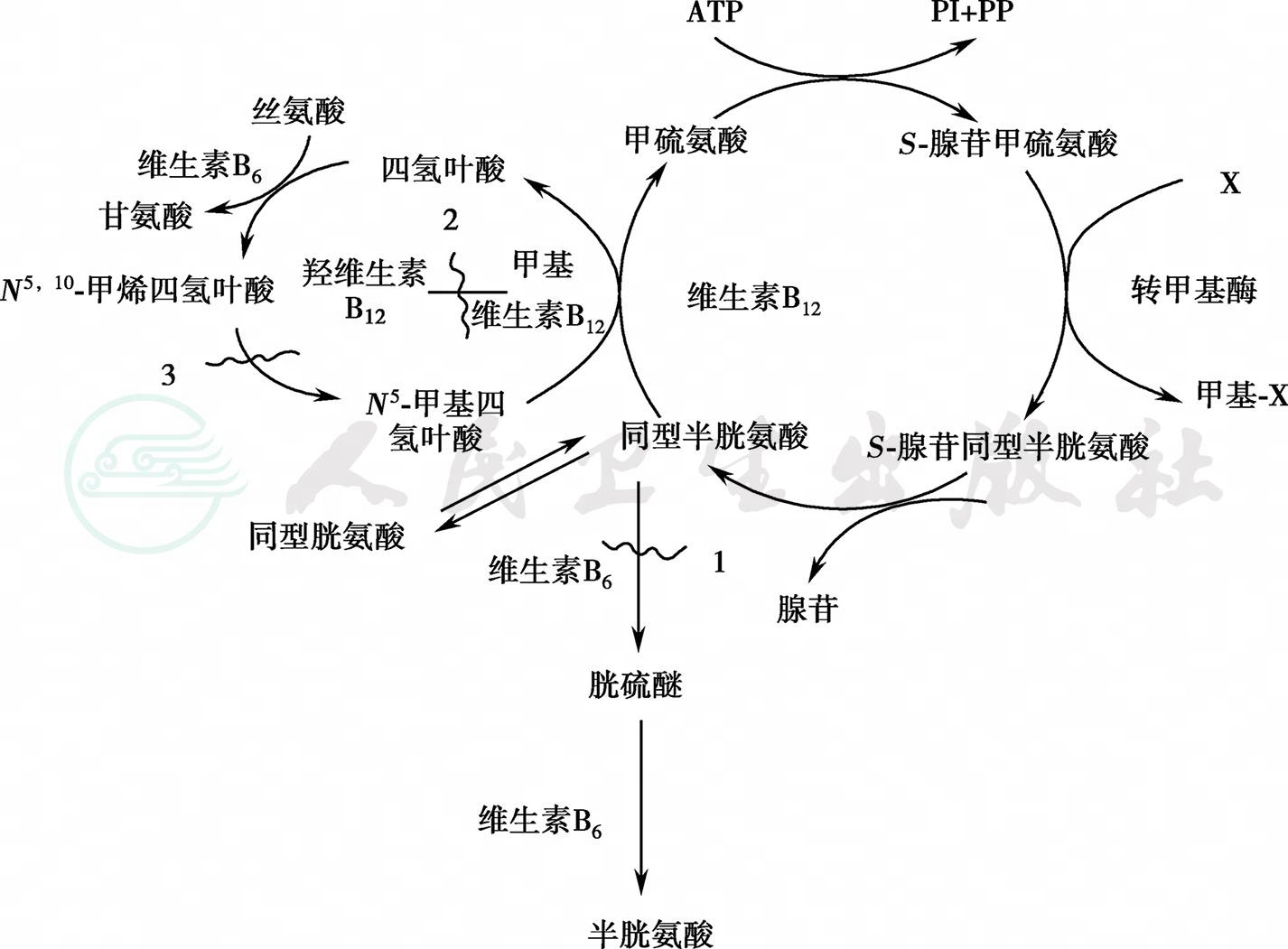
图1甲硫氨酸代谢和同型胱氨酸尿症发病机制
1.同型胱氨酸尿症,合成酶型;2.同型胱氨酸尿症,甲基转移酶型;3.同型胱氨酸尿症,还原酶型。
引自:实用新生儿重症医学.第1版.ISBN:978-7-117-36408-9.主编:
2.甲基四氢叶酸-高半胱氨酸甲基转移酶缺乏型(甲基转移酶型)
此型患者的甲基转移酶酶蛋白本身的活性并未降低,而是辅酶(维生素B12)缺乏。正常时,同型半胱氨酸经过甲基化作用可形成甲硫氨酸,这种转化是在甲基转移酶催化下进行的,所需辅酶是维生素B12的活化型,即甲基维生素B12。本型是由于体内维生素B12代谢异常,不能将体内吸收的维生素B12在细胞内转化为活性型有辅酶功能的维生素B12所致。
3.N5,10-甲烯四氢叶酸还原酶缺乏型(还原酶型)
此型的功能是催化N5,10-甲烯四氢叶酸还原为N5-甲基四氢叶酸。后者可为同型半胱氨酸经甲基化而转变为甲硫氨酸的反应中提供甲基。该酶缺乏时,不能形成足够的N5-甲基四氢叶酸,所以可引起同型胱氨酸甲基化不足而沉积于体内,同时出现同型胱氨酸尿症。
同型胱氨酸尿症为单基因病,属常染色体隐性遗传性代谢病。根据最新GeneReviews资料,编码胱硫醚合成酶的基因为CBS;编码甲硫氨酸合成酶(cblC型、cblD型、cblF型、cblG型、cblJ型)的基因分别为MMACHC、MMADHC、LMBRD1、MTR、ABCD4; 编 码N5,10-甲烯四氢叶酸还原酶的基因为MTHFR;各亚型的OMIM及其编码基因一般情况见表1。CBS基因至今已发现130余种突变,多数突变无共性;MTHFR基因已发现24种突变;cblC缺陷型是钴胺素代谢障碍中最常见类型,其编码基因为MMACHC,已发现近50种突变;cblD型存在cblD-1和cblD-2两种变异型:MMADHC基因编码的蛋白N-末端区域发生改变,即cblD-1变异型,引起单纯同型半胱氨酸血症;C-末端区域发生改变,即cblD-2变异型,引起单纯甲基丙二酸血症;外显子5、8或内含子7发生突变可导致经典型cblD表型,即甲基丙二酸血症合并同型半胱氨酸血症。编码cblE、cblF和cblG的基因MTRR、LMBRD1和MTR突变报道较少。cblJ型为新近报道,其编码基因为ABCD4。
表1同型胱氨酸尿症编码基因的一般情况
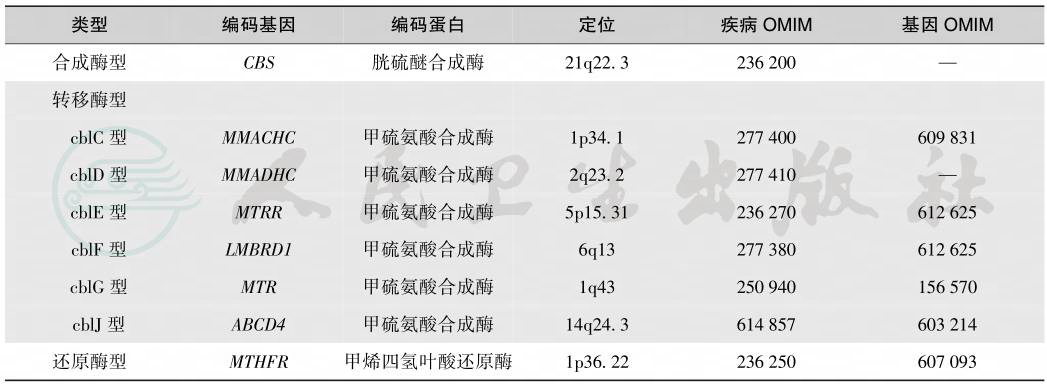
引自:实用新生儿重症医学.第1版.ISBN:978-7-117-36408-9.主编:
1.常规检查
甲基转移酶型有巨幼细胞贫血,血中叶酸水平升高。
2.尿有机酸、血氨基酸谱分析
血和尿中同型胱氨酸过高;合成酶缺乏型血中甲硫氨酸升高;转移酶缺乏型和还原酶缺乏型血、尿中胱硫醚浓度增多。
3.酶活性测定
肝活检测定酶活性,也可用皮肤成纤维细胞测定酶活性;产前诊断可测羊水细胞的酶活性。
4.基因突变分析
应用一代或NGS测序技术对编码胱硫醚合成酶、甲硫氨酸合成酶和N5,10-甲烯四氢叶酸还原酶基因进行突变分析,可确诊同型胱氨酸尿症并可明确其基因分型。
5.其他
X线检查可发现前述骨质改变情况。眼部可发现晶状体脱位、近视、青光眼及视网膜脱离等表现;血管钙化,血管造影血管内膜呈条纹波浪状外观,体循环和肺血管阻塞(血栓形成和栓塞)。
1.合成酶缺乏型
应试用大剂量维生素B6(100~500mg/d)和低甲硫氨酸饮食治疗:对维生素B6敏感者,可加用叶酸和维生素B12,这三种维生素结合可以降低同型半胱氨酸水平,并提供临床益处;对于大剂量维生素B6完全无效者,应补充胱氨酸,加用甜菜碱。测量同型胱氨酸水平可用于监测治疗效果。合成酶缺乏型预后较差,若不经治疗,多于20~30岁死于血管栓塞并发症。
2.还原酶缺乏型
不需要限制蛋白质摄入量,通过口服甜菜碱3~6g/d(最大量可达10g/d)和亚叶酸0.5~1.5mg/(kg•d),可获得良好的控制。还原酶缺乏型预后尚可,可存活至成年。
3.甲基转移酶缺乏型
需使用羟钴胺或甲钴胺治疗,每次肌内注射1mg,每周2~3次,患者巨幼细胞贫血可纠正,但神经系统损伤很难恢复。甲基转移酶缺乏型预后大多不良,可早期死于反复感染。









