


 收藏
收藏 已收藏
已收藏1927年,Guido Fanconi首次报道了3例全血细胞减少合并多发性先天畸形儿童患者。不久,Uehlinger报道了1例相似的患者,伴有拇指和肾脏的畸形。1931年,Naegeli将其命名为先天性再生障碍性贫血,也称为范科尼贫血(Fanconi anemia,FA)。
随后逐渐认识到FA是一种罕见的多系统异常遗传病,主要表现为骨髓造血衰竭、皮肤改变[咖啡牛奶斑(café au lait spots)]、躯体畸形(somatic malformations)[身材矮小、小头畸形、小眼裂,多指(趾)或并指(趾),单侧肾缺如等]、肿瘤易感(cancer prone disorder)[血液系统肿瘤,如急性髓细胞性白血病(acute myelogenous leukemia,AML)和实体瘤,如女性乳腺癌、肺癌、头颈部上皮癌]等,对烷化剂和促炎细胞因子敏感。FA是最常见的先天性骨髓衰竭综合征[congenital(inherited)bone marrow failure syndrome,CBMFS或IBMFS]。遗传方式多为常染色体隐性遗传,2%的患者为X染色体隐性遗传,其基因携带率约为1/300,发病率约为1/1 000万~3/1 000万,儿童发病率约为1/360 000。
由于DNA损伤修复蛋白基因的突变而引起的基因组不稳定是FA发生的关键机制。DNA修复蛋白是非致瘤性的,DNA修复对维持基因组完整性极为重要,DNA修复的任何异常都会使正常细胞分裂过程中其他基因发生突变。
FA是对DNA损伤药物高度敏感的一组疾病。目前报道与FA相关的突变基因有20个,但公认的致病基因为16个(FANCA、FANCB、FANCC、BRCA2、FANCD2、FANCE、FANCF、FANCG、FANCI、BRIP1、FANCL、FANCM*、PALB2、SLX4、ERCC4和UBE2T)。另外一些基因会导致染色体脆性综合征,并伴有FA相关的畸形,但一般不发生骨髓造血衰竭,被称为FA-样基因(RAD51C、RAD51、BRCA1、FAAP100、FAAP24、FAAP20、CENPS、CENPX、BOD1L1、UHRF1、USP1、UAF1和FAN1)。临床上64%为FANCA突变、14%为FANCC突变、9%为FANCG突变。FANCB、BRCA2、FANCD2、FANCE和FANCF突变约13%。FANCA突变最常见,且倾向于晚期发生骨髓造血衰竭,FANCC和FANCG致病基因导致的临床表现更为严重,可能需要较早期进行造血干细胞移植。在一个家族中鉴定FA基因的突变,有助于进行产前诊断和围着床期的遗传学诊断及FA携带者的诊断。
1.血常规
外周血以血小板减少伴有粒细胞减少和/或红细胞减少的两系或三系血细胞减少。常为大红细胞,网织红细胞绝对值减少。白细胞总数明显减少,主要为粒细胞减少。血小板计数减少,血小板的体积正常。
2.骨髓检查
髂骨为骨髓穿刺首选部位。骨髓穿刺涂片外观可见油滴。染色涂片可见骨髓增生活跃或增生减低,有核细胞总数明显减少,粒系和红系细胞比例减少,细胞形态正常;淋巴细胞比例增高,网状细胞、浆细胞和肥大细胞等非造血细胞增多;脂肪较多,巨核细胞减少;骨髓小粒造血细胞成分减少。染色体核型正常,干(祖)细胞培养粒细胞单核细胞集落生成单位(CFU-GM)、爆裂型红细胞集落生成单位(BFU-E)、混合集落生成单位(CFU-MIX)生长减低。
3.骨髓活体组织检查
骨髓有核细胞增生减低,脂肪髓(见图1D),纤维染色阴性,巨核细胞减少。
4.血液学以外的异常
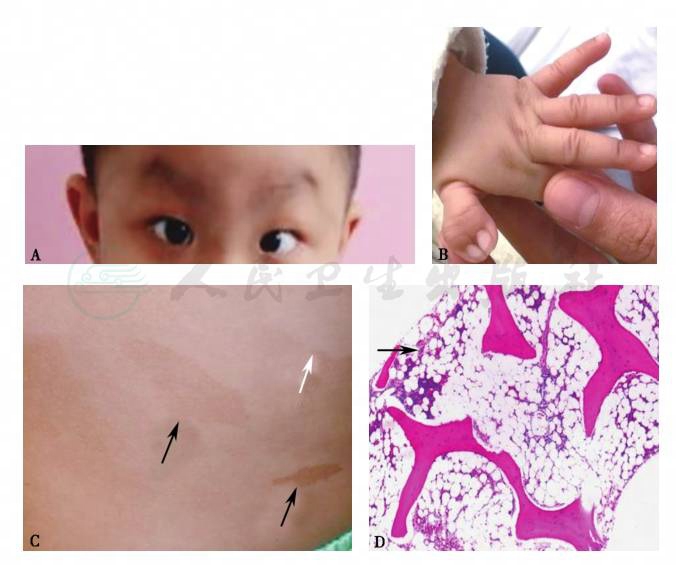
图1 范科尼贫血的发育畸形、皮肤改变及骨髓病理显示的脂肪化(临床病例)
A.眼距增宽、内斜视、皮肤色素沉着;B.并指畸形;C.白色箭头为牛奶斑(色素脱失),黑色箭头为咖啡斑(色素沉着);D.骨髓病理显示脂肪髓(箭头区域)
患者可有肝脏、肾脏、心脏、泌尿道、胃肠道、听觉和视觉,以及内分泌(甲状腺、糖耐量、垂体功能、青春期后的性腺)功能异常,骨骼发育畸形。出现多系统发育异常时,需要做脑垂体磁共振成像和磁共振血管造影除外垂体病变和烟雾病。另外,一些癌症发生与FA相关,特别是口腔癌。
5.抗碱血红蛋白
胎儿血红蛋白(HbF)可正常或增高。
6.染色体不稳定试验
FA细胞对DNA交联剂(DNA cross-linking agent)如丝裂霉素(MMC)、二环氧丁烷(DEB)异常敏感。目前普遍用于临床检测的是MMC试验。部分FA患者的血液存在镶嵌现象(mosaicism),这些患者建立的B淋巴细胞系常为MMC/DEB耐药株,但其成纤维细胞对MMC敏感,可用于诊断。
7.二代测序
包括家系验证的二代测序技术有助于FA的诊断和分型。二代测序可表现为纯合突变和复合杂合突变。
由于不同的FA基因突变均呈异质性,很难通过基因测序鉴别FA路径,Shimamura基于FA信号转导途径的研究进展建立了一种通过FANCD2蛋白疫印迹或免疫荧光,筛查单泛素化(monoubiquitinated)FANCD2蛋白,快速诊断FA和亚型的筛查方法。对于一个最初染色体断裂实验阴性但临床怀疑为FA的患者,免疫印迹可能有助于鉴别嵌合体状态,继而做进一步检查。
FA的平均发病年龄是6.5岁,但诊断年龄为0~50岁,一些非典型表现(如骨髓造血衰竭但无躯体发育异常)的患者往往到成人时才诊断明确。
具有典型的血液学异常(单系或三系血细胞减少)和躯体畸形等临床表现者可以进行临床诊断。但无躯体畸形等典型临床表现者需进行MMC试验检查阳性、二代测序的纯合突变和复合杂合突变有助于诊断和分型。现介绍国际FA研究基金会2008年提出的临床及实验室诊断依据。
1.诊断要点和诊断标准
(1)主要条件
①有阳性家族史;②骨髓增生减低;③特征性先天畸形;④自发性染色体断裂;⑤儿童MDS;⑥儿童急性髓细胞性白血病;⑦对化(放)疗异常敏感;⑧伴乳腺或其他肿瘤家族史。
(2)次要条件
①血细胞减少的家族史;②不能用维生素B12和叶酸缺乏解释的大细胞性贫血;③非肝炎性和非酒精性肝炎的肝脏肿瘤;④<30岁卵巢衰竭;⑤<5岁脑肿瘤;⑥<4岁肾母细胞瘤;⑦不能解释的HbF增高;⑧男/女不孕症。
(3)实验室诊断依据
①染色体断裂实验阳性;②FANCD2单泛素化缺陷;③基因检测发现相关基因突变;④HbF可正常/增高;⑤流式细胞分析发现G2/M期阻滞。
2.鉴别诊断
范科尼贫血的鉴别诊断比较复杂,在临床实际工作中需要与非范科尼贫血性骨髓衰竭综合征、染色体不稳定综合征、伴有先天畸形和智力发育障碍的遗传性疾病,以及非范科尼贫血性青少年白血病及肿瘤等疾病进行鉴别。
(1)非范科尼贫血性骨髓衰竭综合征
各种原因引起的贫血、血小板减少、遗传与非遗传性骨髓衰竭综合征是需与范科尼贫血鉴别的最常见疾病。包括先天性纯红细胞再生障碍、先天性角化不良、重型先天性中性粒细胞缺乏症、舒-戴二氏综合征、血小板减少伴桡骨缺失综合征、Pearson综合征等。但是这组疾病当中患者形体及智力发育障碍、肿瘤并发少见,并且染色体断裂实验结果为阴性,以资鉴别。在少见情况下,Seckel综合征可伴有形体和智力发育障碍、贫血及染色体断裂实验阳性,但分子技术检测可帮助鉴别。
(2)染色体不稳定综合征
染色体不稳定综合征(chromosome instability syndrome)包括很多种疾病,其中以Bloom综合征和共济失调毛细血管括张症(ataxia telangiectasia)为常见。尽管这两种疾病会有与范科尼贫血类似的临床表现,但是偶尔也可表现为染色体断裂实验阳性,也可并发特别类型的肿瘤。但是这两组疾病不伴有骨髓衰竭,同时也不并发MDS与AML。
(3)先天畸形和智力发育障碍(intellectual developmental disorder)的遗传性疾病
这组患者表现为先天性畸形,智力发育障碍,并可见到单一或多脏器功能障碍,但是这组疾病一般不伴有骨髓衰竭,并且染色体断裂实验为阴性。
(4)非范科尼贫血性青少年白血病及肿瘤
这组患者当中不伴有躯体畸形及智力异常,可有骨髓衰竭但一般染色体断裂实验为阴性,但如果3个月内患者曾接受放疗与化疗,会有假阳性。
诊断后的血液学评估对治疗选择极为重要。①血常规检查正常、无细胞形态学及细胞遗传学异常的FA患者每6个月评估1次血常规,每年评估1次细胞形态学及细胞遗传学。②诊断时轻度血细胞减少、无细胞形态学及细胞遗传学异常者每3个月评估1次血常规,每6个月评估1次骨髓形态学和细胞遗传学。③诊断时中度血细胞减少、无造血衰竭或预后不良的细胞遗传学异常(+ 1q、−20q、−11q、−5q、−Y)者,每6~8周复查1次血细胞,每3~4个月复查1次骨髓形态学和细胞遗传学。①②③三种情形一旦监测到血细胞减少进展,有条件者应选择造血干细胞移植,若无合适供者,无不良预后细胞遗传学异常者进行雄激素治疗。④诊断时严重的血细胞减少(原始细胞< 10%)或不良的细胞遗传学异常(−7q、+ 3q、复杂异常,RUNX1突变)或BRCA2/FANCD1突变者,直接进行造血干细胞移植(诊断后治疗选择流程见图2)。
FA的治疗主要针对其血液学改变,以及危及生命的各种并发症。
1.雄激素和皮质类固醇激素治疗
当FA患者发生全血细胞减少时的治疗主要是雄激素和支持治疗。雄激素增加促红细胞生成素(erythropoietin,EPO)的产生,刺激红系干细胞,从而提高血红蛋白水平。约75%的患者雄激素治疗有效,雄激素起效最早的表现是出现大红细胞以及HbF水平增加,开始治疗后2~3个月血红蛋白开始上升,随后血小板计数上升,最后中性粒细胞上升,要确定雄激素是否有效至少要坚持用药6个月。有效时间几个月至20年不等。几乎所有的患者停用雄激素后复发,仅少数治疗时小于12岁的患者,在青春期时可停止治疗而不复发。最终许多患者对所用的雄激素耐药,换用另外一种雄激素少部分患者可能有效。雄激素的应用延长了患者的生存期,但一些患者可能出现肿瘤等晚期并发症。
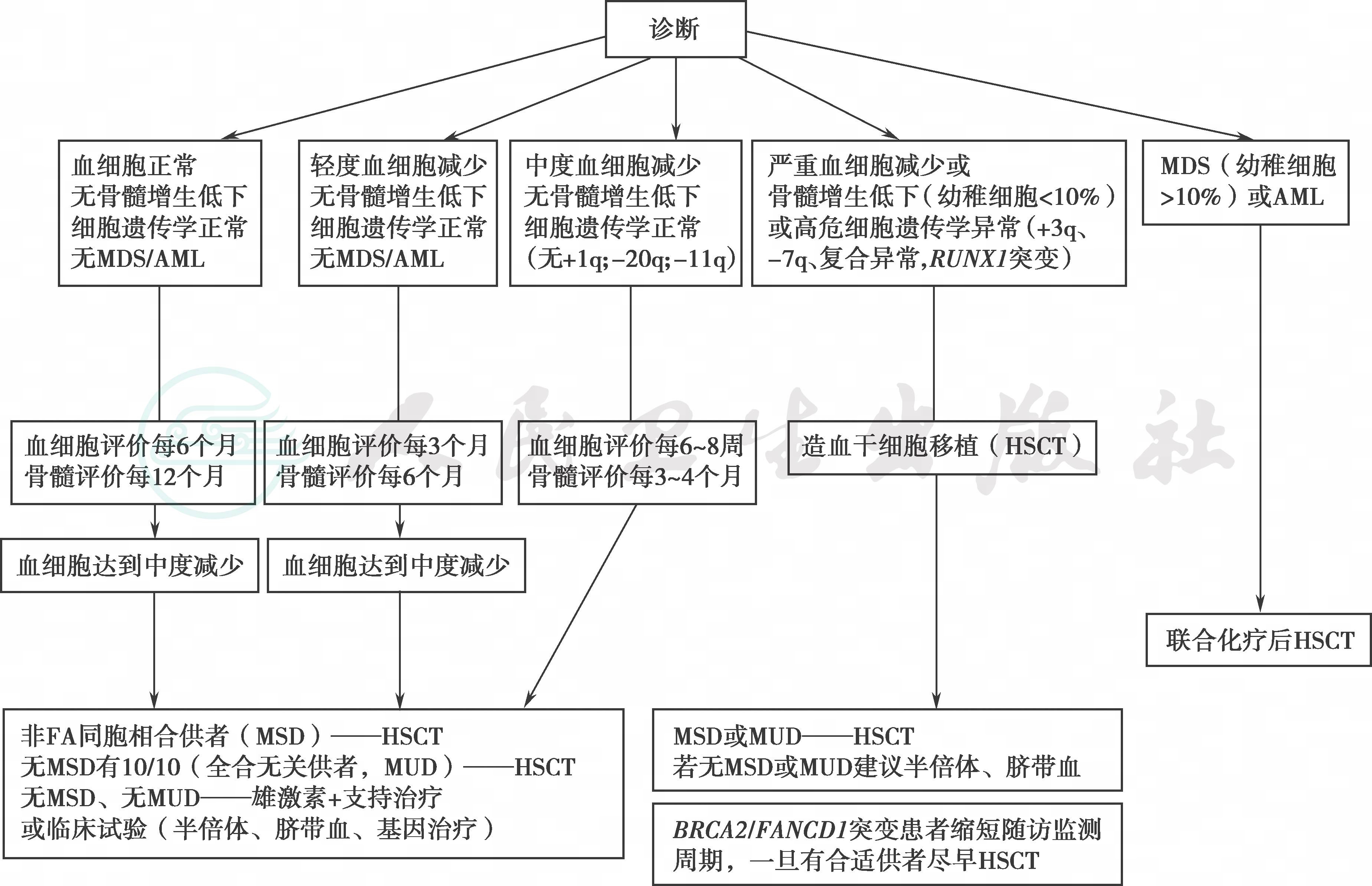
图2 范科尼贫血诊断后治疗流程选择
MDS:骨髓增生异常综合征;AML:急性髓细胞性白血病
单独应用雄激素与雄激素加皮质类固醇激素的疗效相同,但一般推荐联合治疗,皮质类固醇激素引起的生长迟缓可抵消雄激素生长加速的副作用,也可以通过降低血管的通透性减少出血。最常用的雄激素是司坦唑醇,口服2~5mg/(kg·d);联合泼尼松5~10mg,隔日1次口服。为降低肝脏毒性可用雄激素注射剂苯丙酸诺龙每周1~2mg/kg,肌内注射。为预防血肿,可用冰袋冷敷和按压。
雄激素的副作用有妇女男性化、多毛症以及声音变粗,外生殖器肥大,痤疮,情绪不稳,水、钠潴留,体重增加,肌肉发达,由于骨骼成熟加速致骨骺过早融合,最终导致身材矮小。在这些副作用中,部分在雄激素减量或停用后消失。比较严重的副作用包括肝大、胆汁淤积性黄疸和肝功能中转氨酶水平上升,但这些是可逆的。最严重的问题是肝紫癜、肝腺瘤和肝细胞癌,但这些在雄激素治疗停止后也能恢复。接受雄激素治疗的患者需定期进行肝生化检查和超声检查,治疗有效的患者可逐渐减量但不能停药。但有些患者雄激素可停用,这些患者可能有血液系统镶嵌现象,其“正常”干细胞有选择性造血优势。
2.细胞因子
造血生长因子如粒细胞集落刺激因子(G-CSF)和粒细胞-巨噬细胞集落刺激因子(GM-CSF)能改善造血,特别是中性粒细胞减少的患者,能增加中性粒细胞绝对值,仅少数患者血红蛋白和血小板计数增加。可与雄激素联合应用或用于雄激素治疗无效的患者。然而,这些因子的应用也能使肿瘤易感的患者发生白血病或促使向MDS或7号染色体单体演化,因此仅用于严重中性粒细胞减少的患者,不能用于有克隆性细胞遗传学异常的患者,并注意监测外周血细胞计数,定期行骨髓检查和骨髓细胞遗传学检查,一旦发现异常应停用。
3.造血干细胞移植
造血干细胞移植是唯一能治愈FA患者的措施,也可以预防白血病的发生。有健康HLA相合的同胞供者采用异基因造血干细胞移植,2年生存率可达到66%;无HLA相合的同胞供者可选择HLA相合的无关供者或不匹配的家族成员,但移植效果很差,2年生存率仅29%。HLA相合的同胞脐带血移植已有成功的报道,优化预处理方案的无关全相合脐带血移植已有成功报道。
由于FA患者对放疗和预处理方案药物如环磷酰胺超敏,可发生严重的黏膜炎伴有肠道吸收障碍和出血,液体潴留,心力衰竭和出血性膀胱炎。减少环磷酰胺的剂量到20mg/kg,分4天给药,加5Gy的胸腹部放疗,这个方案的累积生存率大约是70%。应用氟达拉宾进行预处理,代替放疗取得了更好的疗效。尽管骨髓移植是一种有效的治疗措施,但化疗和放疗增加了发生第二肿瘤的危险(尤其是头颈部肿瘤)。
4.基因治疗
FA前体细胞和干细胞的基因转导可以从遗传学上纠正所有系统的造血细胞异常,恢复正常的持续造血。在FA-A和FA-C患者的体外试验中均已获得成功,是目前FA基因治疗的依据所在。生物技术的不断发展有望用于临床。
5.其他治疗
FA患者部分病例需要支持治疗。有出血的患者,可用6-氨基己酸0.1g/kg,每6小时服用1次。有可能需要移植治疗的患者,应输注过滤了白细胞的血液制品或辐照血液制品,避免输注来自家族成员的血液制品,以减少移植时移植物抗宿主病。避免接触可抑制骨髓造血的药品和化学物质。血小板减少的患者避免应用影响血小板功能的药物。FA继发AML者治疗困难,预后差。由于DNA修复缺陷,对化疗敏感性增加,因此化疗相关毒性增加,化疗剂量应减少。
FA是一种累及多系统的疾病,移植虽然可以重建骨髓造血,但会增加肿瘤易感性,移植治疗该类疾病的远期并发症及转归尚需得到关注,建立登记网络势在必行。尽管发现了近20个致病基因,但其发病机制仍有待于进一步研究,在此基础上的基因治疗才有望实现。









