


 收藏
收藏 已收藏
已收藏英文名称 :Gitelman syndrome
Gitelman综合征(Gitelman syndrome,GS)是由编码噻嗪类利尿剂敏感的钠氯协同转运蛋白(NCC)的SLC12A3基因突变所致的一种常染色体隐性遗传病。同大多数转运蛋白病一样,GS的发现得益于20世纪下半叶临床生化时代的到来,随着电解质检查成为常规,许多毫无关联的多系统受累可以用某种或某几种电解质异常来解释。1966年美国医生Gitelman等人报道了3例(2例为姐妹)临床表现相似的患者,主要表现为发作性肌无力和弛缓性瘫痪,实验室检查提示低血钾、低血镁和代谢性碱中毒,伴有血浆肾素水平升高,命名为Gitelman综合征。为了确定致病部位,给予这3例患者7天限钾(或限镁)饮食。健康人通过降低尿钾和尿镁排泄,维持血钾和血镁水平正常。但这3例患者尿钾和尿镁水平并不减少,进而将该病定位在肾脏失盐。因为该病与另一种钠钾氯协同转运蛋白2(NKCC2)相关转运蛋白或调节蛋白异常导致的失盐性肾病巴特综合征(Bartter syndrome,BS)有相似的临床表现,很长时间内GS一直被认为是BS的一个亚型。直到1988年,Puschelt等人首次观察到GS患者对作用于远端小管的噻嗪类利尿剂反应不好,其氯排泄量显著低于健康人群,首次将GS病变部位定位到了远端小管,而不是BS受累的髓袢升支粗段。但真正从分子机制上将GS与BS区分开是在1996年,Simon等人克隆出位于染色体(16q13)上编码肾脏远端小管NCC的SLC12A3基因,证实该基因突变是导致GS的原因,并在体外(爪蟾卵母细胞)证实了不同SLC12A3突变的致病性。2007年Colussi等人以基因诊断为“金标准”,验证了简化氢氯噻嗪试验对于评估患者NCC功能、临床诊断GS的价值。北京协和医院肾内科2009年建立了中国人氢氯噻嗪试验的正常值,并首次证实其与基因诊断GS“金标准”相比,敏感性和特异性均接近95%,不仅为临床鉴别GS和BS提供了便捷、安全和经济的方法,还可用于不同原因导致NCC功能障碍的定位诊断和损伤程度定量评估(图1)。
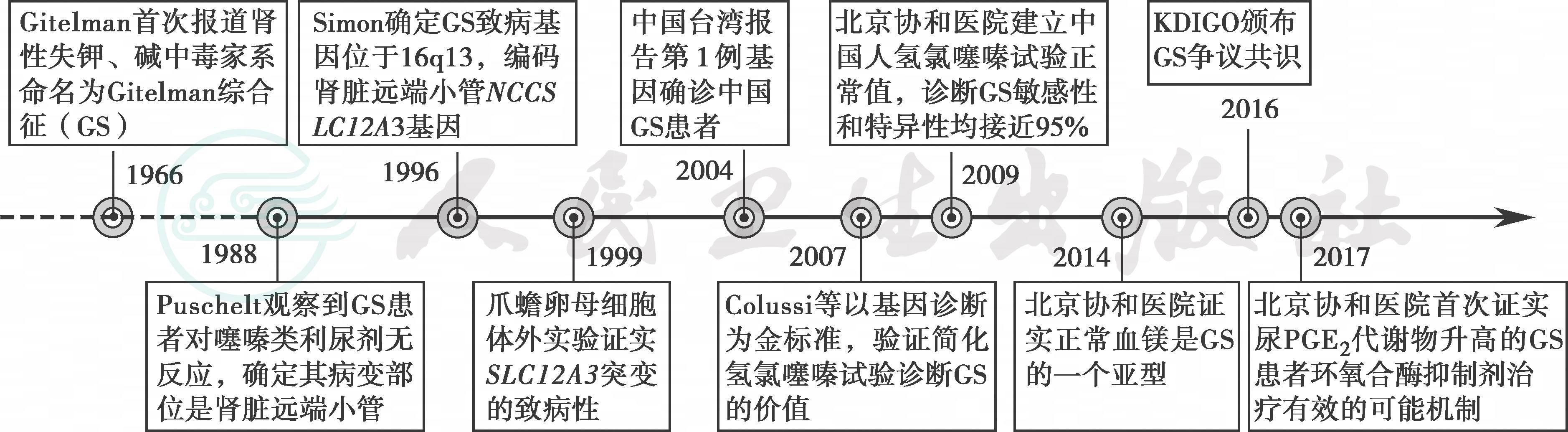
图1 Gitelman综合征的认识过程
GS是最常见的遗传性肾小管疾病之一,患病率约为1/40 000~10/40 000,在亚洲人群中可能更高,全世界已发现超过500种SLC12A3基因突变。在生理情况下,NCC位于肾脏远端小管上皮细胞的管腔侧,参与重吸收肾小球滤过液中约5%~10%氯离子和钠离子,是机体维持水电解质平衡的一道重要防线。当基因突变导致NCC结构和/或功能障碍时,氯离子和钠离子从远端肾小管重吸收减少,继发肾脏重吸收水减少、肾素-血管紧张素-醛固酮系统(RAAS)活化和肾性失钾,高尿镁和低尿钙继发于肾小管上镁和钙转运蛋白功能的异常。随着基因诊断“金标准”用于临床,传统的“低血镁和低尿钙”来鉴别GS和其他失盐性肾病的敏感性和特异性均低于80%,北京协和医院在近百例GS队列中证实正常血镁比例约为20%,可能是GS的一个亚型。该型患者临床表现相对轻,血镁水平与NCC功能损伤程度负相关。肾脏活检证实,本组患者远端小管上负责镁离子转运的瞬时受体电位阳离子通道亚家族M成员6(TRPM6)表达水平高于低血镁的患者,为GS正常血镁亚型提供了结构依据。
GS的临床诊断主要依赖家族史(常染色体隐性遗传)、临床表现和血生化检验,还需要做心电图评估有无心律失常和QT间期延长等表现。确诊依靠基因诊断,氯离子清除试验可明确NCC是否受累和受累的程度,也可在没有条件开展基因检测的单位作为重要的鉴别诊断手段,GS的诊断流程见图2。
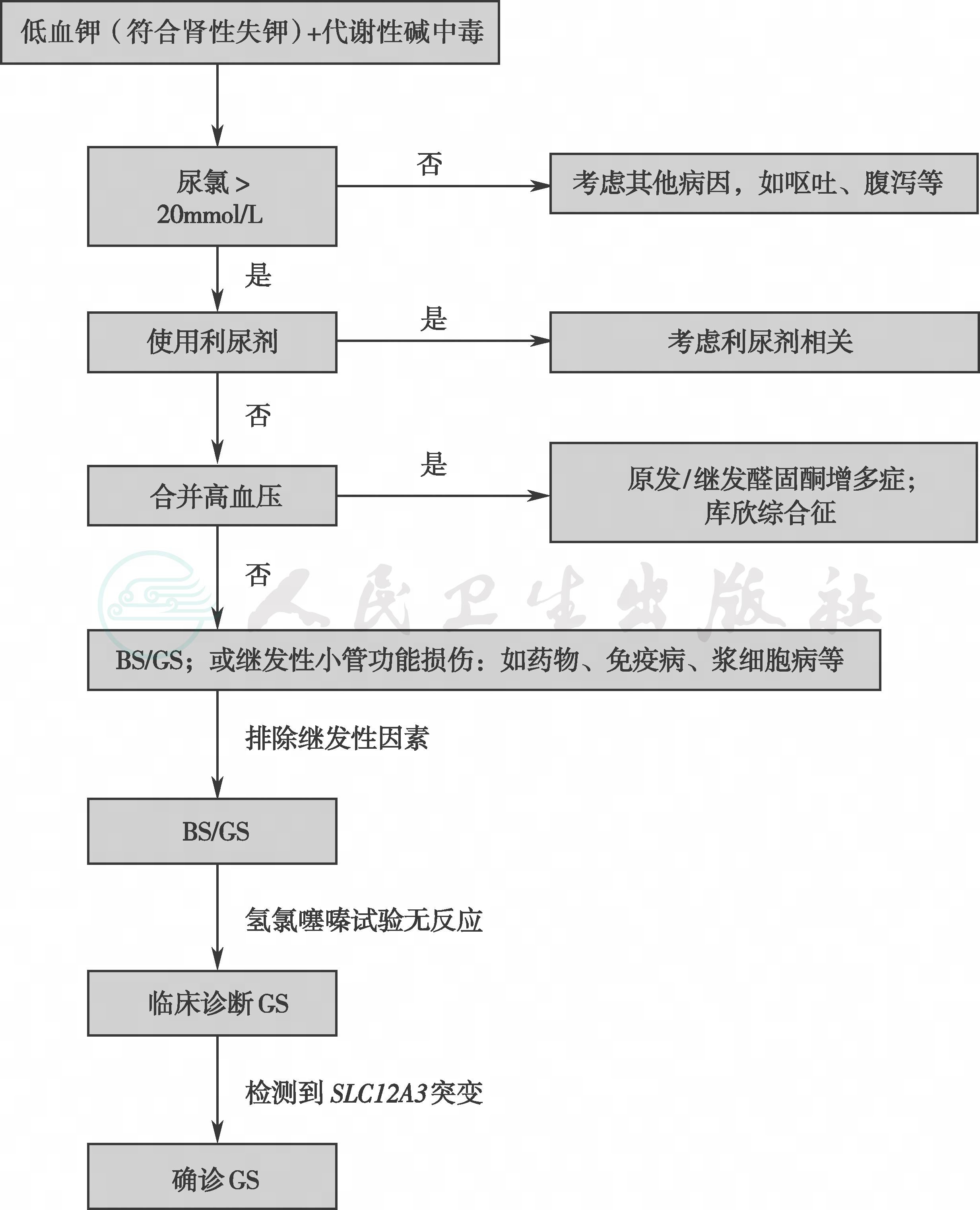
图2 Gitelman综合征诊断流程
GS:Gitelman综合征;BS:Bartter综合征
基因检测是诊断“金标准”,检测到SLC12A3纯合突变或双杂合突变可确诊,单杂合突变的患者需结合临床。如检测到的SLC12A3突变为未报告突变,如何再做突变致病性分析呢?根据美国医学遗传学与基因组学学会(ACMG)2015年发布的基因变异致病性解读指南可对突变进行分类,如遇到无法明确分类的情况,体外功能实验可为确定突变致病性提供证据,常用非洲爪蟾卵母细胞作为载体来完成,将突变和野生型NCC的cRNA注射进入卵母细胞,观察膜上NCC蛋白的表达或者磷酸化蛋白水平,随后分析其摄取22Na水平与野生型的差异,进而确定其NCC功能损伤的程度。
氯离子清除试验原理是给疑诊GS或者BS患者小剂量氢氯噻嗪或呋塞米,观察氯离子清除率数的变化程度(△FECl),GS患者对氢氯噻嗪没有反应,而BS患者对呋塞米没有反应,进而可以从临床上鉴别二者。目前临床采用的简化氯离子清除试验操作较为简便易行,并且成本低,但因试验前一天需停止补钾,试验中需监测血压和心率,警惕进一步降低血钾的风险。
Gitelman综合征暂无根治疗法,但合理的治疗和自我管理,可以达到有效缓解症状、提高生活质量、避免严重并发症的目标。患者可以与健康人一样生长发育、婚育、学习、工作和生活。目前治疗方法包括食物和药物替代治疗、基于发病机制的治疗和一些探索性的治疗。
1.钾和镁替代治疗
推荐高盐饮食,进食富含钾和镁的食物,其次通过口服含钾、含镁药物予以补充,紧急或严重情况下可静脉输注。需要注意的是,钾补充应以氯化钾为主,枸橼酸钾会加重代谢性碱中毒。2017年改善全球肾脏病预后组织(KDIGO)专家争议共识建议血钾和血镁治疗目标分别为3.0mmol/L和0.6mmol/L。
2.基于Gitelman综合征机制的治疗
包括容量不足继发RAAS活化和远端小管液钠含量增高促进钠钾交换两方面。可以选用的药物包括:①抑制RAAS活化的药物,包括血管紧张素转换酶抑制剂(ACEI)、血管紧张素Ⅱ受体拮抗剂(ARB)、醛固酮受体拮抗剂(如螺内酯、依普利酮),以及环氧合酶(COX)抑制剂如吲哚美辛等;②阻断钠-钾离子交换机制的药物,如阿米洛利。这些药物的使用有助于减少补钾药物的剂量,改善低钾相关症状,需要注意的是RAAS抑制剂导致的低血压、螺内酯导致男性乳腺发育、COX抑制剂导致的肾功能(肾小球滤过率)降低等副作用。其中争议较多的是COX抑制剂(吲哚美辛),其有效治疗GS的病例报道并不少,但一直以来难以解释其机制。既往文献在小样本缺乏基因诊断的GS患者中发现COX代谢产物前列腺素E2(PGE2)并不高,北京协和医院近期在基因确诊的GS患者中,观察到男性患者尿中PGE2代谢产物(PGEM)高于女性患者,尿液PGEM水平高的患者临床表现更重,首次提出如果根据尿PGEM水平选择COX活化患者,开展COX抑制剂靶向试验,有助于提高疗效,避免不必要的不良反应。
3.其他探索性治疗
有文献报道,终末期肾病接受肾移植的GS患者,获得了痊愈。随着基因编辑技术的进展,通过远端小管特异性标志物的启动子靶向引导,对NCC异常进行基因治疗,GS痊愈的可能并非遥不可期。









