


 收藏
收藏 已收藏
已收藏英文名称 :Alport syndrome
中文别名 :奥尔波特综合征;遗传性肾炎;眼-耳-肾综合征
Alport综合征(即奥尔波特综合征,Alport syndrome,AS),又称遗传性肾炎、眼-耳-肾综合征,是一种并不少见、遗传方式多样,以血尿、蛋白尿、进行性肾衰竭伴感音神经性耳聋、眼部病变为主要临床表现的遗传性基底膜病。1902年Guthrie首次报道了血尿家系,1927年Alport第一个将家族性血尿与耳聋联系起来并在一个患者中还提到了眼部病变,因此后来将临床表现为血尿为主的肾炎、耳聋和眼部损害的疾病命名为Alport综合征。20世纪70年代早期,随着电镜的创立和发展,AS肾小球基底膜超微结构下厚薄不均改变被不同实验室发现并报道(Kinoshita等,1969年;Spear和Slusser,1972年;Hinglais等,1972年;Churg和Sherman,1973年),因此研究者考虑AS是一种基底膜疾病。直到现在,肾小球基底膜超微特征性改变仍是AS重要的诊断依据之一。1980—1990年,一系列免疫组化的研究表明AS的基底膜病变与基底膜主要成分Ⅳ型胶原异常有关。1988年Atkin首先将AS相关基因定位于X染色体,1990年Barker等成功克隆到编码Ⅳ型胶原蛋白α5链的基因(COL4A5基因),并在X伴性遗传AS患者中发现了第一个COL4A5基因突变。由于Ⅳ型胶原蛋白α3、α4、α5链形成一个Ⅳ型胶原单体,因此随后的研究发现编码Ⅳ型胶原蛋白α3链基因(COL4A3)和α4链基因(COL4A4)与常染色体隐性遗传AS有关,随着近年来基因检测技术的广泛开展,发现更多的COL4A3和COL4A4基因单杂合突变与常染色体显性遗传AS相关。基于上述研究发现近年来有研究者称AS为“Ⅳ型胶原相关肾病”,但目前这一说法尚未得到广泛认可。2018年国际AS研究专家组会议建议将Ⅳ型胶原α3~α5链分子异常导致的所有疾病统称为AS。
AS发病与基底膜主要框架结构成分之一——Ⅳ型胶原(type Ⅳ collagen)亚单位α3~α5链编码基因COL4A3、COL4A4、COL4A5突变有关,其中,COL4A3、COL4A4位于2号常染色体(2q36),COL4A5位于X染色体(Xq22)。家系调查及一代基因测序研究结果提示AS遗传方式有3种:X伴性遗传AS(X-linked AS,XLAS)(OMIM#301050),约占80%~85%,最为常见;常染色体隐性遗传AS(autosomal recessive AS,ARAS)(OMIM #203780),约占15%;常染色体显性遗传AS(autosomal dominant AS,ADAS)(OMIM#104200)最为少见。但随着二代及其他基因检测技术的快速发展及临床推广,发现ADAS远远多于ARAS,且发现一些更为特殊的遗传方式如双基因突变(表1),其中COL4A3/COL4A4基因反式突变类似常染色体隐性遗传方式,COL4A3/COL4A4基因顺式突变类似常染色体显性遗传方式,但基因COL4A5和基因COL4A3/COL4A4双基因突变不符合任何一种孟德尔遗传方式。
(一)肾组织常规病理检查
1.光镜无特异性
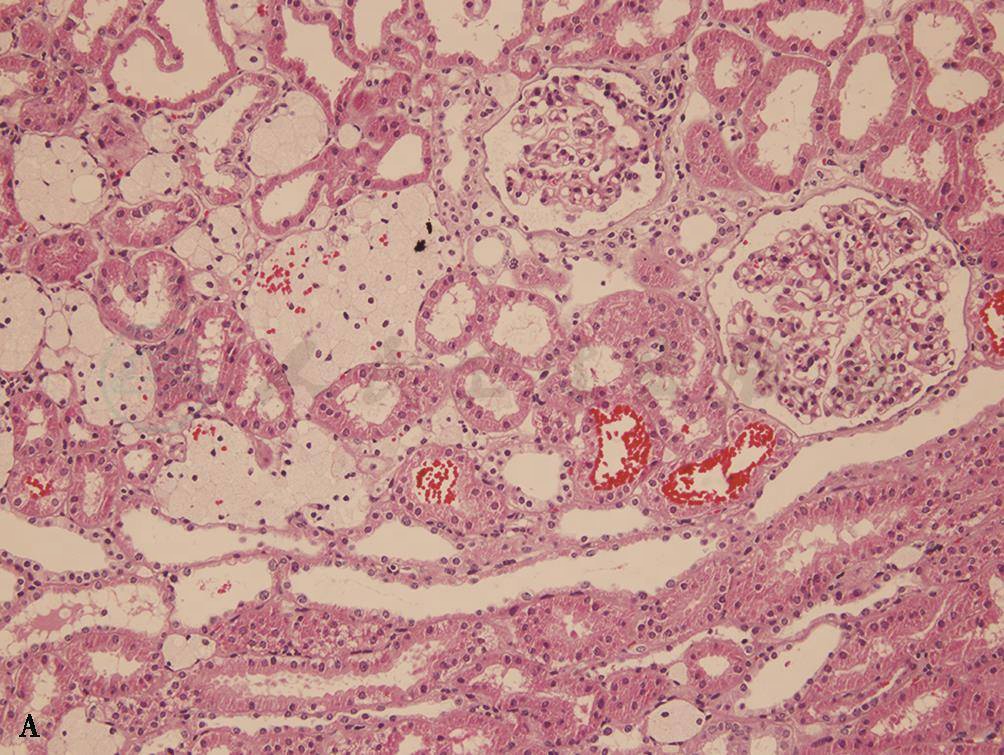
疾病早期或5岁之前,肾小球和肾血管基本正常,5岁以上患者可出现系膜和毛细血管袢改变,光镜下表现为轻微病变、局灶性节段性肾小球硬化、弥漫性系膜增生等。约40%肾组织标本可有间质泡沫细胞(图1A),此改变不具诊断意义,但若发现间质泡沫细胞,应注意有无AS可能,尤其临床无肾病综合征表现者。
2.免疫荧光(IF)多为阴性
少数标本系膜区、毛细血管壁可有IgA、IgG、IgM、C3、C4等局灶节段或弥漫沉积,有报道及上海交通大学医学院附属瑞金医院资料均显示极少数患者可有IgA在系膜区弥漫沉积,甚至被误诊为IgA肾病。
3.电镜改变多种多样
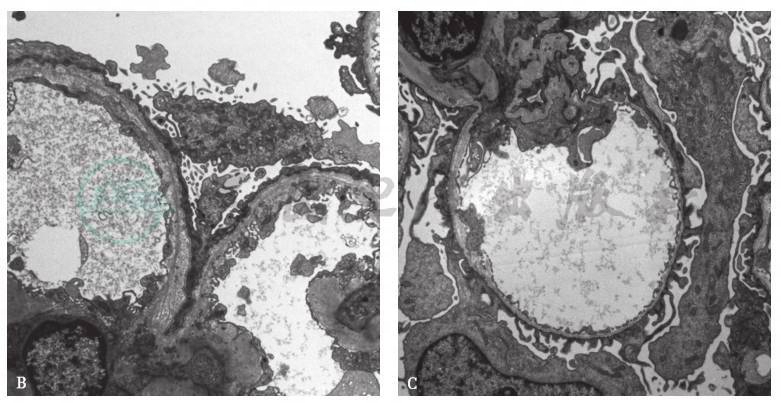
典型者呈弥漫肾小球基底膜(GBM)厚薄不均、分层、网篮样改变(图1B),极少数可见GBM断裂,多数XLAS男性、ARAS患者及少数XLAS女性表现为典型改变,部分儿童、XLAS女性和ADAS患者可表现为弥漫GBM变薄(图1C)。
A
B C
图1 Alport综合征肾脏病理表现
A.光镜下肾间质泡沫细胞(HE,200 ×);B.电镜示肾小球基底膜(GBM)厚薄不均、分层、网篮样改变(18 000 ×);C.电镜示 GBM弥漫变薄(18 000 ×)
(二)皮肤及肾组织Ⅳ型胶原不同α链间接免疫荧光检测
在正常情况下,抗α3、α4(Ⅳ)链抗体在GBM、远端肾小管基底膜(dTBM)、抗α5(Ⅳ)链在GBM、包氏囊(BC)、dTBM、表皮基底膜(EBM)上沉积,免疫荧光呈连续线样。而α3~α5(Ⅳ)链在XLAS、ARAS患者肾组织和皮肤沉积见表2、图2和图3,约75% XLAS男性和50% XLAS女性及部分ARAS患者可发现以上改变。该检测方法具有重要诊断意义,且有助于AS遗传方式的确定。
(一)家族史
表2 Ⅳ型胶原不同α链在正常肾组织及AS患者中免疫荧光检测结果
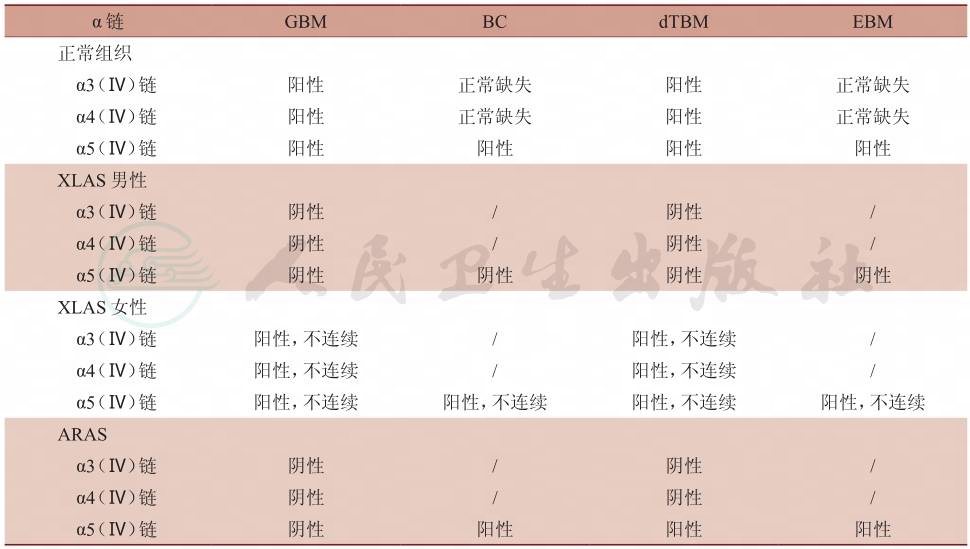
GBM:肾小球基底膜;BC:包氏囊;dTBM:远端肾小管基底膜;EBM:表皮基底膜;/:正常缺失
除通过详细询问家族史并绘制家系图外,尽量对先证者父母乃至家系中成员进行尿检及肾功能筛查。基于AS中新发突变比例大于10%,即使没有肾脏相关的家族史,疑似患者仍不能排除AS诊断,需基因和/或Ⅳ型胶原不同α链检测明确。
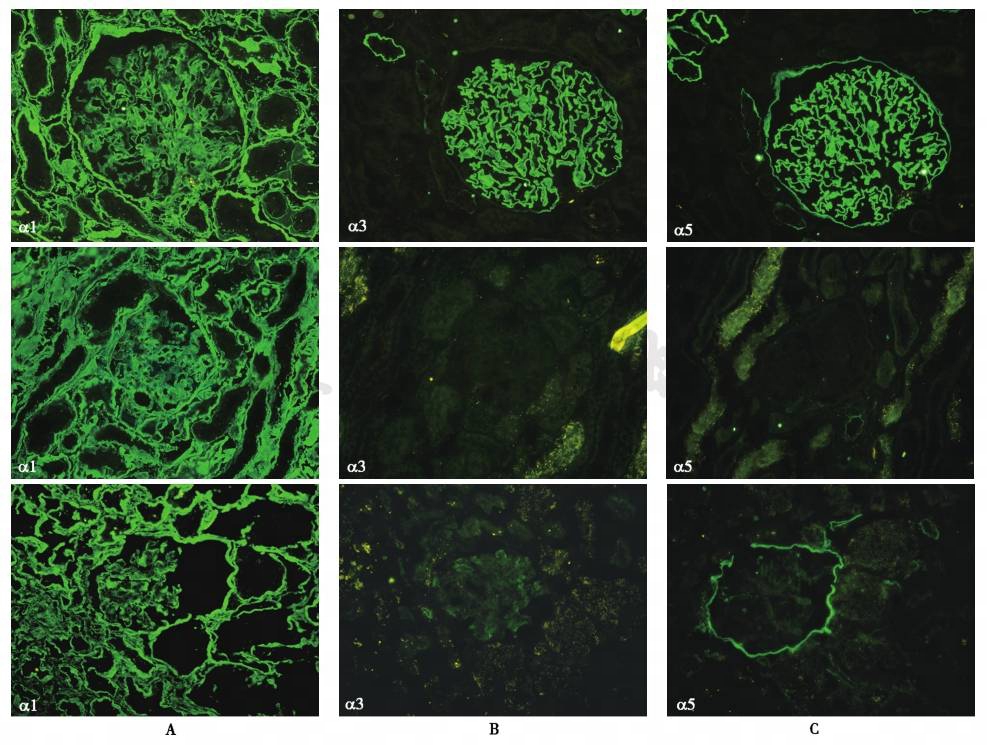
图2 Alport综合征肾组织Ⅳ型胶原不同α链检测结果
A.α1(Ⅳ)链;B.α3(Ⅳ)链;C.α5(Ⅳ)链(免疫荧光染色,400 ×)
(二)诊断标准
AS诊断必须结合临床表现、电镜、家族史、Ⅳ型胶原不同α链检测结果等综合判断(图4)。即主要表现为持续性肾小球性血尿或血尿伴蛋白尿的患者,有AS家族史或排除其他原因的血尿、肾衰竭家族史或有听力及眼部病变,则疑诊AS。进一步检查,符合以下标准任意一条即可确认AS:组织(肾组织和/或皮肤组织)基底膜α(Ⅳ)链免疫荧光染色异常;电镜示GBM致密层撕裂分层;COL4An基因分析明确存在致病性突变。建议对每一个AS家系通过基因检测进行遗传型诊断,便于对先证者进行预后评估,且对其家系进行遗传咨询(图4)。
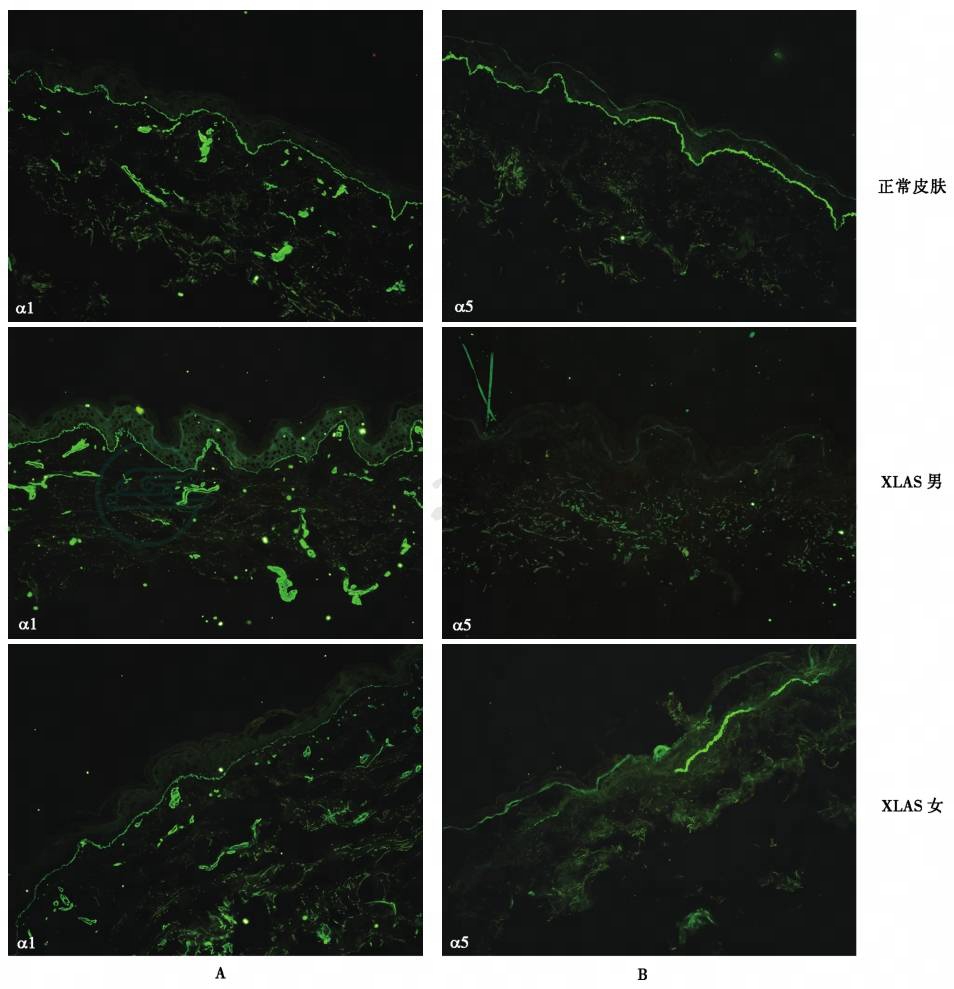
图3 Alport综合征皮肤组织Ⅳ型胶原不同α链检测结果
A.α1(Ⅳ)链;B.α5(Ⅳ)链(免疫荧光染色,200×)
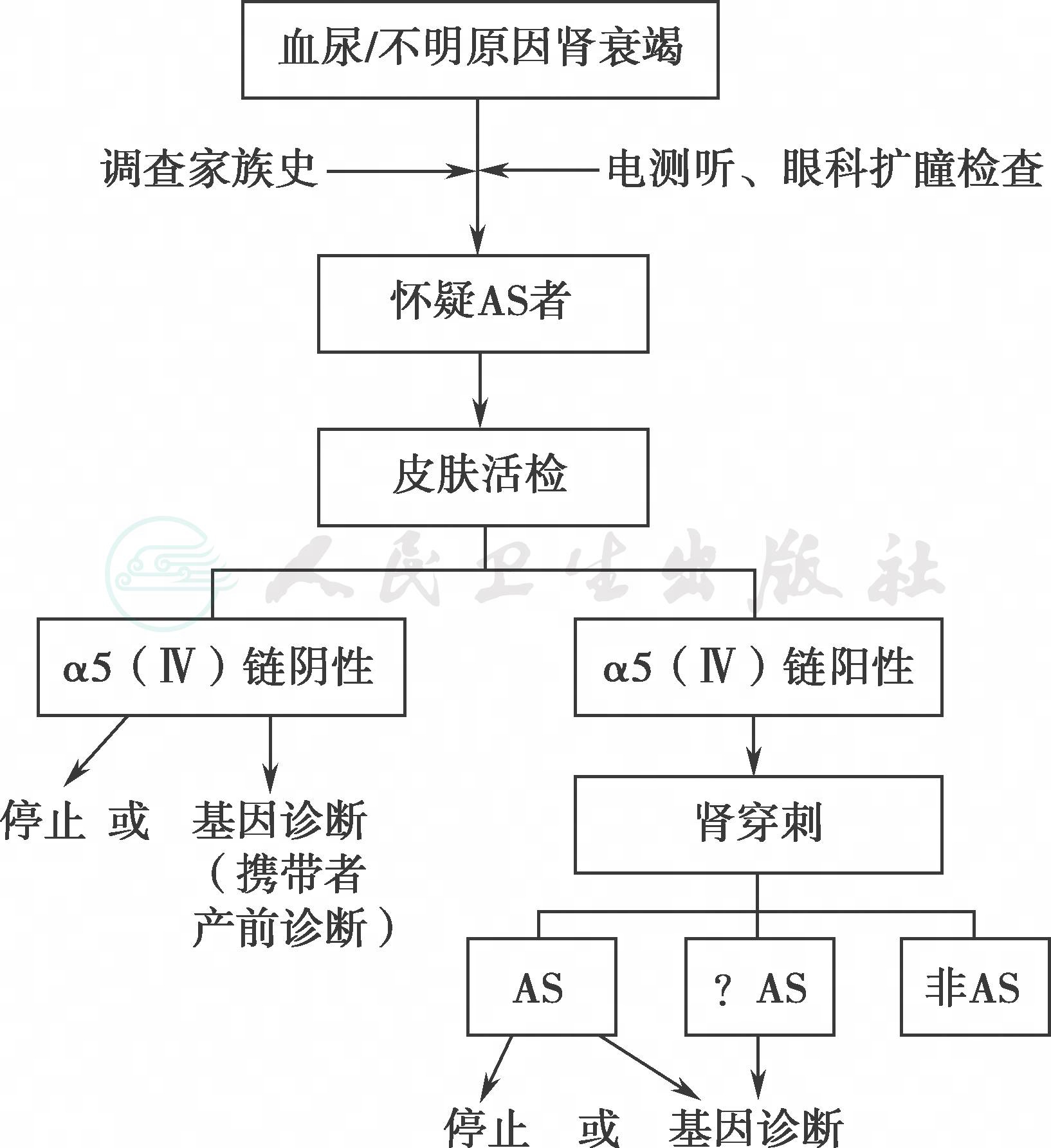
图4 Alport综合征(AS)患者的诊断思路
(一)治疗目的
控制尿蛋白,减轻肾小管上皮细胞损伤,抑制肾间质纤维化,延缓进展至肾衰竭的速度,维持肾功能。
(二)药物治疗建议
Alport综合征专家诊治建议将治疗药物分为一线和二线用药,其中一线治疗应用ACEI(雷米普利、依那普利等);二线治疗应用ARB(氯沙坦、厄贝沙坦、缬沙坦等)和醛固酮受体拮抗剂(螺内酯),早期应用(微量白蛋白尿期)可减少尿蛋白并延缓肾功能进展。但对有生育需求的育龄期妇女,应谨慎使用ACEI和ARB。建议接受上述药物治疗的AS患者注意监测血钾及血清肌酐,当患者短期内血清肌酐升高超过30%,或者出现直立性低血压时应进行相应的减量。大多数医生在临床实践中出于对不良反应的考虑,在血清肌酐大于265mmol/L时不使用肾素-血管紧张素-醛固酮系统阻断剂。此外,既往有报道指出环孢素A能够有效降低AS蛋白尿,但也有报道环孢素A治疗效果不明显,且肾毒性作用方面存在争议,从而限制其应用。激素和其他免疫抑制剂对AS进程有弊无利。
(三)肾脏替代治疗建议
进展至ESRD的AS患者需要肾脏替代治疗,包括透析和肾移植。总的来说,AS患者有很好的移植效果。最近的研究表明,AS患者肾移植后20年的存活率为70.2%,移植肾的存活率为46.8%,明显优于其他肾脏疾病的肾移植效果。但需要注意到的是,约3%~4%患者可发生移植后抗GBM抗体性肾炎,此类患者再移植效果差。
(四)对症治疗
在目前缺乏有效治疗的情况下,对症治疗仍非常重要:①减少蛋白摄入;②控制高血压;③纠正贫血、水电解质酸碱紊乱;④积极查找和去除感染灶;⑤避免肾毒性药物。
(五)患者管理和遗传咨询
一旦诊断AS,患者要严密随访,进行合理的遗传咨询和饮食指导,并尽量完善家系筛查。建议患者每3个月行尿液相关检查,包括尿常规、尿蛋白/肌酐、24小时尿蛋白定量等。同时建议,每6~12个月进行肾功能评估。对于有生育要求的患者进行相关遗传宣教,给予专业的指导。
(六)新的治疗探索
研究者一直在探索新的治疗,转基因小鼠实验显示一些干预可能获得有益的效果,这些干预包括尝试逆转基因缺陷如干细胞移植、逆转肾小球细胞的信号通路异常、阻滞TGF-β1介导的纤维化等。抗microRNA-21治疗转基因Alport小鼠,显示其可减轻肾小球炎症,并可影响肾纤维化途径,目前一个Ⅱ期临床试验正在18岁以上、GFR在45~90ml/min患者中开展。此外甲基巴多索隆(bardoxolone methyl)治疗AS的临床研究亦在进行中。









