


 收藏
收藏 已收藏
已收藏英文名称 :progressive familial intrahepatic cholestasis
自20世纪50年代,相继报道了多例累及婴幼儿和儿童的肝内胆汁淤积病例。其中一部分为肝内胆道闭锁病例,一部分为良性复发性肝内胆汁淤积病例,另有一部分病例病程呈进行性、组织学表现为小叶间胆管减少,被归为家族性肝内胆汁淤积。
1969年Clayton等报道了发生在阿米什家族中的致命性家族性肝内胆汁淤积症。在4个相互关联的近亲家族中出现7例肝内胆汁淤积患者,均自婴儿起病,临床表现为皮肤瘙痒及波动性黄疸,伴吸收不良,体检均有肝脾大,肝功能异常的特点为胆红素升高及碱性磷酸酶升高。在这7例患儿中,有5例在17个月~8岁期间死亡。作者将其命名为Byler病,即现今的进行性家族性肝内胆汁淤积症(progressive familial intrahepatic cholestasis,PFIC)-1型(OMIM #211600)。
此后相继有非Byler家系PFIC患者的报道,人们逐渐认识到PFIC是一组异质性疾病。1996年Bourke等报道了非Byler家系的8例PFIC患者,其临床特点与Byler病相似,称之为Byler综合征,即现今的PFIC-2型(OMIM #601847)。此后相继定义了PFIC-3型(OMIM #602347)及其他新的亚型,不同亚型的致病基因不同。
由于属于罕见疾病,PFIC的发病率无确切报道,出生时PFIC-1型和PFIC-2型的发病率为1/100 000~1/50 000,发病与性别无关。文献报道10%~15%儿童胆汁淤积性疾病归因于PFIC,10%~15%儿童肝移植归因于PFIC。2018年5月,PFIC被国家卫生健康委员会等五部门联合制定的《第一批罕见病目录》收录。
PFIC是一组常染色体隐性遗传性疾病。因基因突变导致胆汁排泌障碍,发生肝内胆汁淤积,最终可发展为肝衰竭。
根据其致病基因不同,该疾病主要分为3型,包括PFIC-1型、PFIC-2型和PFIC-3型。
(一)PFIC-1型
由ATP8B1基因突变引起,ATP8B1位于常染色体18q21-q22,该基因编码P型ATP酶——家族性肝内胆汁淤积1(familial intrahepatic cholestasis 1,FIC1)缺陷。FIC1蛋白位于肝细胞毛细胆管膜,它负责调节氨基磷脂转入细胞内,维持毛细胆管膜双分子层内膜高浓度的氨基磷脂。其功能异常可间接干扰胆管胆汁酸分泌。
(二)PFIC-2型
由三磷酸腺苷结合盒B亚家族成员11(ATP-binding cassette sub-family B member 11,ABCB11)基因突变引起,ABCB11基因位于常染色体2q24,该基因编码胆盐排泄泵(bile salt export pump,BSEP),该蛋白是肝细胞毛细胆管膜胆盐转运蛋白,属ABC转运蛋白家族成员,BSEP蛋白缺陷导致胆盐分泌减低,胆流减少,从而使肝细胞内胆盐积聚,造成损伤。
(三)PFIC-3型
由三磷酸腺苷结合盒B亚家族成员4(ATP-binding cassette sub-family B member 4,ABCB4)基因突变引起,编码多药耐药糖蛋白(multidrug resistance associated protein 3,MDR3)。MDR3主要在肝细胞毛细胆管膜表达,其功能产物磷脂酰胆碱转出酶调节磷脂从双分子层向外移动,是磷脂转运器。其缺陷导致胆固醇结晶,胆汁结石形成增加,阻塞小胆道。
近年来,二代测序和全外显子测序技术的发展使我们能发现更多与PFIC相关的基因缺陷。例如,新近将位于染色体9q21上的紧密连接蛋白2(tight junction protein 2,TJP2)基因变异所导致的PFIC定义为PFIC-4型(OMIM #615878);将位于染色体12q23上的编码法尼醇X受体的NR1H4基因变异所导致的PFIC定义为PFIC-5型(OMIM #617049)。
(一)实验室检查
三种类型PFIC血清学检查均呈胆汁淤积性改变,表现为血结合胆红素、碱性磷酸酶及胆汁酸呈不同程度增高,血胆固醇多正常。血清γ-谷氨酰转移酶(GGT)持续升高有助于鉴别PFIC-3与PFIC-1和PFIC-2,PFIC-3型血清GGT增高,而PFIC-1型和PFIC-2型GGT正常。
(二)影像学检查
磁共振胰胆管成像(MRCP)或腹部超声等检查可观察肝内外胆管,PFIC一般无肝内外胆管异常改变。
(三)病理学检查
病理学检查有助于PFIC的诊断和鉴别诊断。
1.PFIC-1型
肝组织最特征表现为电镜下粗颗粒状胆汁,称为“Byler胆汁”,部分肝细胞可按管状模式排列,形成腺泡样假“玫瑰花结”。其他非特异性表现包括肝细胞空泡变性、轻微炎性细胞浸润、毛细胆管内胆汁淤积、汇管区轻微小管增生和纤维化等,肝巨细胞形成不明显。
2.PFIC-2型
肝组织病理特征性的表现在于明显的肝巨细胞的形成,电镜下胆汁呈细丝状、细颗粒状或无定形状,微绒毛缺失。BSEP免疫组化染色可显示该蛋白表达的缺乏或明显下降。
3.PFIC-3型
肝组织的病理改变类似于肝外胆道闭锁者,有胆管增生和纤维化两个突出表现。增生的胆管被认为是真正的胆管,而不是PFIC-1型患者肝细胞的胆管上皮细胞化生。纤维化程度轻重不一,可以仅仅是汇管区的纤维化,也可以是整个肝组织的广泛纤维化。疾病晚期则表现为胆汁性肝硬化。胆汁淤积程度不一,肝细胞、毛细胆管、各级胆管均可受累。MDR3免疫组化染色可以显示该蛋白在肝脏组织的表达情况。
(四)基因检测
应用DNA测序检测ATP8B1、ABCB11、ABCB4等基因外显子,必要时可采用RT-PCR和测序检测非编码序列和内含子的突变以及剪接错误,或者进行全基因测序。
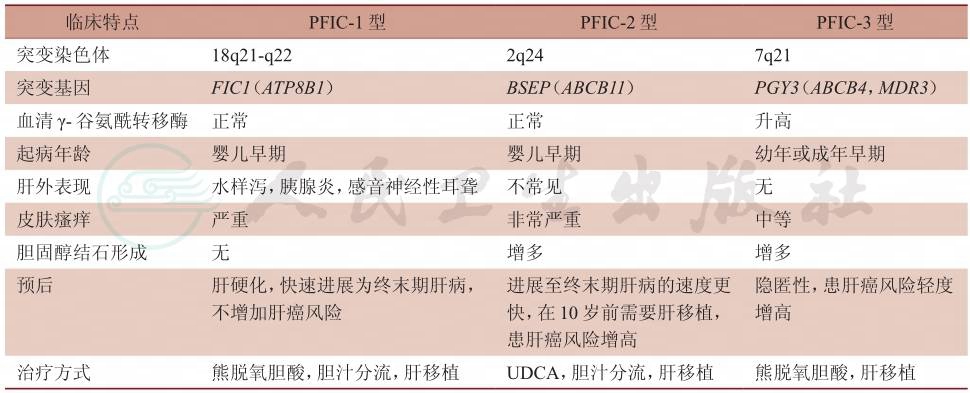
PFIC的诊断依靠临床表现、血生化、胆汁成分分析、肝组织病理学检查以及基因检测等综合判断,并需要排除其他原因所致的胆汁淤积性肝病。各亚型PFIC的临床特点比较见表1。
表1 PFIC各亚型的临床特点比较
PFIC治疗包括对症治疗、药物治疗、外科手术治疗和肝移植。目的是缓解症状,改善营养状态,纠正维生素缺乏以及治疗腹水、食管静脉曲张破裂出血等并发症。
(一)对症治疗
膳食提供中链甘油三酯(又称三酰甘油),改善患儿营养状态。服用脂溶性维生素和水溶性维生素。保证充足的阳光照射和钙摄入。
(二)药物治疗
熊脱氧胆酸(ursodeoxycholic acid,UDCA)对三种类型的PFIC都有效,是所有类型患儿的初始治疗选择,口服剂量为10~30mg/(kg·d)。熊脱氧胆酸可以竞争初级胆汁酸在小肠的重吸收,有效取代其肠肝循环,促进其排出,从而缓解胆汁淤积对肝细胞的损伤。对于PFIC-2型患者,其原发缺陷直接影响胆盐从微管流出,应用熊脱氧胆酸疗效欠佳。对于PFIC-3型患者,熊脱氧胆酸约对2/3的患者有效,但对变异导致无MDR3表达的患者无效。
考来烯胺可以用来缓解胆汁淤积性瘙痒。苯扎贝特和S-腺苷甲硫氨酸的疗效有待于验证。
(三)外科治疗
胆汁分流术是主要术式,包括部分胆汁分流术和回肠旁路手术两大类,部分PFIC-1型和PFIC-2型患者可受益。
(四)肝移植
肝移植是三种类型PFIC患者最为有效、彻底,也是最后考虑的治疗方法。
PFIC是一组以累及婴幼儿为主的常染色体隐性遗传性疾病,其诊断和治疗困难。若能在出现肝纤维化和肝硬化前诊断和积极治疗,可显著降低终末期肝病的发生率和相关死亡率。基因检测技术的迅速发展使我们能更方便、快捷地对可疑PFIC患者进行基因诊断,也使我们能发现更多与PFIC相关的基因缺陷。另外,随着对胆汁酸代谢的深入研究,我们希望能找到新的针对胆汁酸循环/代谢异常的治疗靶点,从而为PFIC提供有前景的治疗药物。









