


 收藏
收藏 已收藏
已收藏家族性高胆固醇血症(familial hypercholesterolemia,FH)是常染色体显性遗传疾病,其高水平的低密度脂蛋白胆固醇(low density lipoproteincholesterol,LDL-C)与早期发生较为严重的冠心病(coronary artery disease,CAD)相关。临床分为纯合与杂合两种类型。杂合患者早发冠心病风险比同年龄段的正常人群增加3~13倍。纯合患者出生即受高胆固醇血症影响,儿童期即可出现全身动脉粥样硬化(atherosclerosis,AS),表现出极端的临床特征:血浆LDL-C为正常人的6~8倍、皮肤多部位黄色瘤和早发冠心病,青少年期可发生心肌梗死甚至死亡。冠心病发生率为正常人群的100倍,自然寿命为20~30岁。
(一)发现历程
1985年诺贝尔生理学或医学奖获得者Goldstein和Brown证实FH分子病理基础是由于低密度脂蛋白受体(low density lipoprotein receptor,LDLR)基因突变所致的受体功能缺陷。此后又发现了载脂蛋白B(lipoprotein B,APOB)、前蛋白转化酶-枯草溶菌素9(proprotein convertase subtilisin/kexin type 9,PCSK9)和低密度脂蛋白受体衔接蛋白1(LDL receptor adaptor protein 1,LDLRAP1)基因突变,其中以LDLR基因突变最为常见。按照基因分型,FH可分为纯合子家族性高胆固醇血症(homozygote familial hypercholesterolemia,HoFH)、复合杂合子家族性高胆固醇血症(compound heterozygote familial hypercholesterolemia)、双重杂合子家族性高胆固醇血症(double heterozygote familial hypercholesterolemia)和杂合子家族性高胆固醇血症(heterozygote familial hypercholesterolemia,HeFH)4种类型,前3类突变均属基因重度缺陷。除单基因突变致病外,部分FH患者为多基因突变的叠加效应所致。
(二)流行病学
纯合FH的患病率约为1/400 000~1/300 000,世界大约有1 000万患者。杂合FH在脂质紊乱性疾病中的发病率较高,在55岁以前发生冠心病的患者中有5%~10%由FH引起,在心肌梗死存活患者中这一比例高达10%。而我国缺乏流行病学资料。
(三)发病机制
基因突变对LDLR功能的影响如下:
1.LDLR
LDLR构成胆固醇代谢经典的途径,它是细胞跨膜糖蛋白,广泛分布于肝、动脉壁平滑肌细胞、肾上腺皮质细胞等,可与含ApoB100的脂蛋白高亲和力结合,介导血中大约70%的低密度脂蛋白(LDL)进入肝细胞内溶酶体降解。LDLR是FH最主要的致病基因,可占所有突变的70%以上。迄今为止英国LDLR基因突变数据库已收录了世界范围内1 700多种突变,其中2/3为点突变或小片段缺失和插入突变,1/3突变为DNA大片段重排。目前国外学者对中国FH患者进行研究,陆续发现了一些中国人群特有的LDLR基因突变位点,除外显子18外,所有外显子均有突变发生。
2.ApoB100
ApoB100是单链糖蛋白,主要在肝脏合成。ApoB100是LDL-C的载脂蛋白,也是LDLR的天然配体。该蛋白基因3 500位及其相邻位点突变可造成LDLR结合结构域的空间构象改变,使其与LDLR的亲和力显著降低,导致LDL-C清除障碍、血清胆固醇升高。目前已经报道有45种突变。1986年首次发现并将该病命名为家族性载脂蛋白B100缺陷症(familial defective apolipoprotein B100,FDB)。APOB100基因突变可占所有FH患者的5%,我国也可见报道。
3.PCSK9
PCSK9是一种蛋白水解酶,能结合并水解LDLR蛋白而减少肝脏对胆固醇的清除,对LDLR有负调控作用,二者互相制约在控制体内胆固醇代谢和稳态中发挥着非常重要的作用。PCSK9基因的功能获得性突变增强了其水解LDLR蛋白的功能,可引起LDLR蛋白减少而循环中LDL-C大幅增高,导致常染色体显性高胆固醇血症3型(autosomal dominant hypercholesterolemia 3,HCHOLA3),该疾病在2003年被法国学者发现,也称FH3,已在多个国家和地区被证实。目前已发现8种突变,我国也可见报道。
4.LDLRAP1
LDLRAP1是LDLR适配器蛋白,其磷酸酪氨酸结合结构域结合于LDLR的胞内部分,促进LDLR与被膜小窝衔接发挥内吞作用。LDLRAP1基因突变可降低LDLR内吞活性,导致常染色体隐性高胆固醇血症(autosomal recessive hypercholesterolemia,ARH),是第4种类型FH。目前已经检测到LDLRAP1基因的9种突变。
5.单核苷酸多态性(SNP)
虽然发现了上述4种相关的致病基因,但仍然不能解释所有临床确诊FH的发病原因。2013年报道指出,英国学者选取与LDL-C水平相关性较强的12个SNP(包括LDLR、PCSK9及ApoB100)建立了加权评分胆固醇遗传风险预测模型,发现与携带致病基因突变的FH患者相比,突变阴性组SNP的评分显著升高,认为多个SNP的累积效应参与了FH患者胆固醇代谢,并提出在未检测到已知基因突变的临床FH患者中,很多是由多基因遗传模式所致。
目前针对HoFH,主要有药物治疗、脂蛋白分离术和肝脏移植,均以降低LDL-C、延缓心脑血管动脉粥样硬化疾病的发病时间、降低死亡率和致残率为主要治疗目标。
根据欧洲动脉粥样硬化学会(EAS)HoFH共识的最新建议:一般成人要求LDL-C < 2.5mmol/L或降低LDL-C至少50%;如果合并有冠心病或糖尿病,则要求将LDL-C控制在1.8mmol/L以下;FH儿童需要合理饮食并从3~10岁开始他汀治疗,建议10岁以后治疗目标为LDL-C < 3.5mmol/L,然而对于HoFH患者很难达到上述目标。
(一)常规治疗
1.他汀类药物
通过竞争性抑制内源性胆固醇合成限速酶、上调LDLR活性而发挥降脂作用,目前仍是FH治疗的首选用药。
2.胆固醇吸收抑制剂依折麦布
选择性抑制肠道对胆固醇的吸收从而降低LDL-C。
3.烟酸类及其衍生物
通过减少脂质的生成和促进其分解而实现调脂作用。
4.胆酸螯合剂
通过在肠道与胆酸结合,阻止肠道对胆酸及胆固醇的吸收,促进其随粪便排泄,可降低血中总胆固醇。
(二)新型药物
PCSK9抑制剂通过抑制PCSK9与LDLR结合而阻断LDLR的降解过程,从而发挥降低LDL-C的作用。Evolocumab是一种全人单克隆免疫球蛋白G2(IgG2),能特异性地与PCSK9结合,在HoFH患者的Ⅲ期临床试验中显示其可降低LDL-C水平达30.9%,目前已被欧盟委员会及美国食品药品监督管理局批准用于治疗成人或12岁以上青少年HoFH患者,剂量为每个月420mg皮下注射。其疗效取决于LDLR基因突变类型,对于LDLR缺失突变的纯合患者无效。
(三)血液净化
血浆脂蛋白置换术可以将血液中LDL-C及ApoB在体外清除后再回输至体内,具体包括免疫吸附、硫酸右旋糖酐纤维素吸附、肝素介导的体外LDL及纤维蛋白原沉淀、全血脂蛋白直接吸附及硫酸右旋糖酐纤维素直接灌注。以上技术可使LDL平均减少60%以上,脂蛋白a下降46%~75%,建议HoFH儿童应于5~8岁开始每周1次血浆脂蛋白置换治疗。长期治疗能使FH患者血浆胆固醇维持在较低水平,皮肤、肌腱黄色瘤消退,心血管并发症降低。
(四)外科手术
肝脏移植术采用正常肝脏植入HoFH患者体内,从而恢复患者对血浆LDL-C的清除能力。对于药物治疗和血浆脂蛋白置换术效果不好或不能耐受的年轻HoFH患者,可考虑肝移植,手术最佳治疗时间应在心血管事件发生之前。
(五)联合治疗
最高耐受剂量的他汀(如阿托伐他汀和瑞舒伐他汀)联合依折麦布能使LDL-C在最初治疗时达标。其他药物如胆酸螯合剂和烟酸也可作为补充治疗。目前,已被批准用于FH治疗的新型药物有PCSK9抑制剂、ApoB反义寡核苷酸药(mipomersen)和微粒体甘油三酯转移蛋白抑制剂(lomitapide)。欧洲动脉粥样硬化学会(EAS)建议HoFH患者的治疗规范如图1所示。
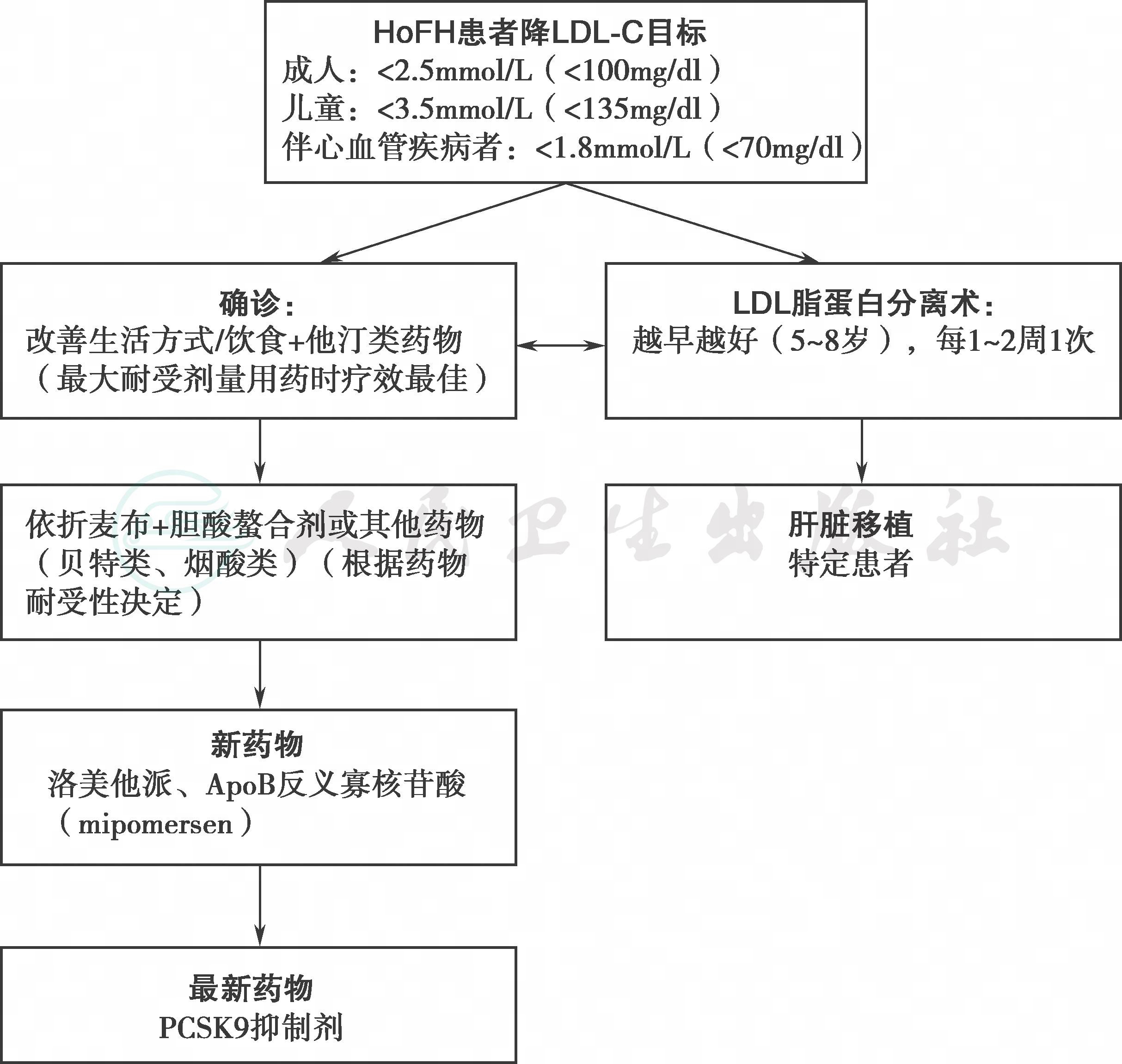
图1 EAS推荐HoFH患者治疗流程









