


 收藏
收藏 已收藏
已收藏英文名称 :hyperinsulinism hypoglycemia
高胰岛素血症性低血糖症(hyperinsulinism hypoglycemia,HH)是胰岛B细胞过量分泌胰岛素或分泌失调所致的以顽固的低血糖为主要临床表现的一组疾病,也是新生儿和婴儿时期持续反复发作低血糖的主要病因。由于婴儿时期神经系统的快速发育,对葡萄糖的需求大,持续、反复发作的低血糖如不能得到及时有效的干预,极易造成永久性脑损害,发生癫痫、脑瘫,甚至死亡。由于胰岛素的作用是促进葡萄糖进入骨骼肌、脂肪组织等胰岛素敏感部位,同时可以抑制糖原分解和糖异生,抑制脂肪酸释放和酮体合成,因此过多分泌的胰岛素导致葡萄糖和酮体功能被抑制,使得循环中维持正常脑活动所需的能量来源(葡萄糖和酮体)减少。据报道,由于诊断延迟和治疗不足,HH患儿的永久性脑损伤风险仍高达25%~50%。
1937年,美国圣路易斯儿童医院的Hartmann和Jaudon对一些低血糖患儿回顾性分析后,提出HH是某些患儿低血糖的原因。Irvine McQuarrie于1953年在美国儿科协会的演讲中首次详细描述了“特发性自发性婴儿低血糖症”。McQuarrie强调由于诊断延迟或治疗不足,患儿遭受不可逆的脑损害,应当引起儿科医生的密切关注。不久后,多伦多的Cochrane等人描述了由高蛋白质饮食或亮氨酸引起的低血糖,后来被称为“蛋白质敏感性低血糖症”或“亮氨酸敏感性低血糖症”。20世纪60年代,随着胰岛素检测技术的开发,“婴儿期特发性低血糖”的病因之一被证实为胰岛素过度分泌。与此同时还发现二氮嗪可以治疗HH;胰腺大部切除术是二氮嗪无反应性HH的儿童控制低血糖症的唯一选择。在20世纪70~80年代,有学者提出胚胎期导管上皮细胞的异常增殖导致了本病的发病,称为“胰岛母细胞增殖症”(nesidioblastosis)。后来的研究否定了这个假说,“胰岛母细胞增殖症”这个名词被摒弃。1995年,首次发现了HH由ATP敏感性钾离子通道基因突变导致。在最近20年中,与HH相关的11种基因被发现,诊断和治疗方法也被极大地改进。
HH可以是具有基因缺陷的先天性高胰岛素血症(congenital hyperinsulinism,CHI),也可以继发于某些风险因素如宫内发育迟缓、围生期窒息、Rh溶血等,某些综合征如Beckwith-Wiedemann综合征、先天性糖基化异常(congenital disorders of glycosylation,CDG)综合征等也与HH有关(表1)。西方国家估计发病率为1/50 000活产婴儿(散发型)。在近亲结婚率高的人群中,估计发病率为1/2 500(家族型)。国内尚缺乏统计资料。遗传方式可为隐性、显性或散发。
目前根据病因不同可将HH分为3类:①先天性高胰岛素血症(CHI);②类先天性高胰岛素血症性低血糖症(congenital HH-mimickers);③后天获得性高胰岛素血症性低血糖症(acquired HH)。各型HH的病因见表1。
表1 高胰岛素血症性低血糖症(HH)的病因分类
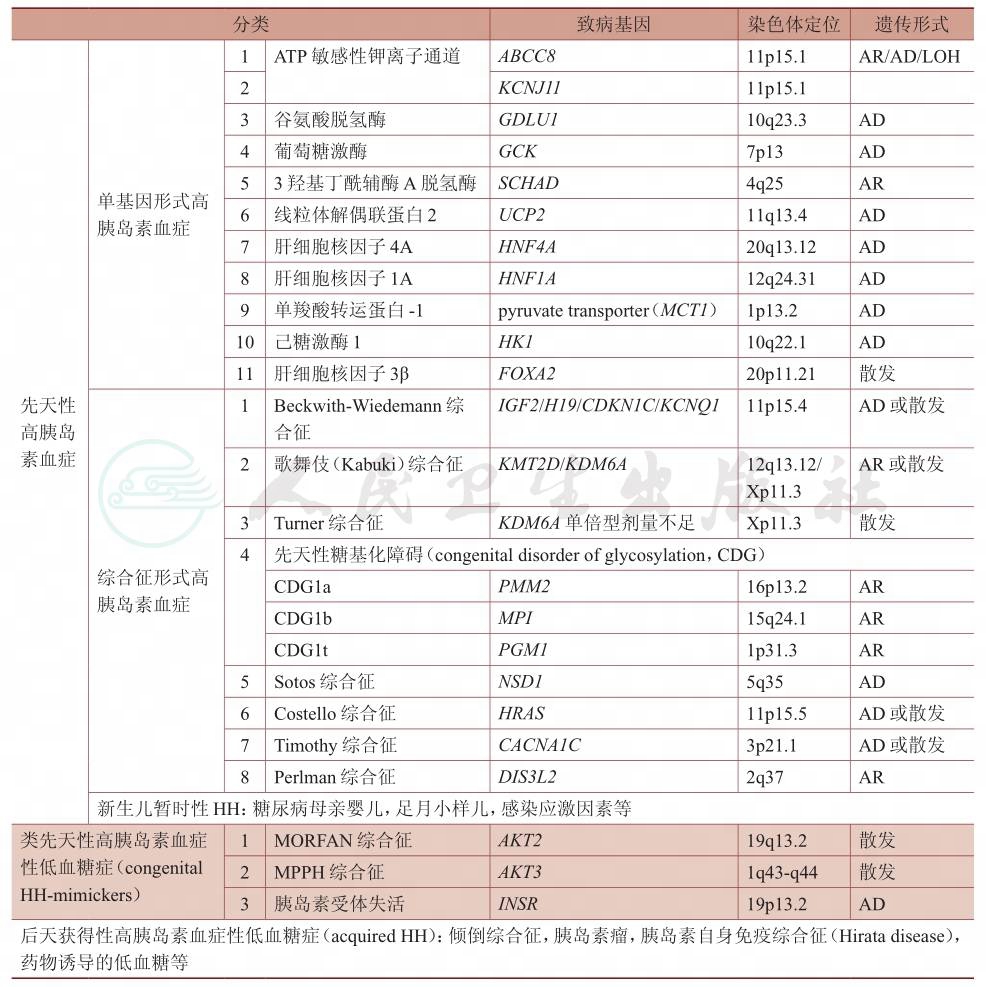
ABCC8:ATP结合盒亚家族C成员8;AKT:丝苏氨酸蛋白激酶;FOXA2:肝细胞核因子3β;GCK:葡萄糖激酶;GDLU:谷氨酸脱氢酶;HK1:己糖激酶1;HNF4A:肝细胞核因子4A;HNF1A:肝细胞核因子1A;INSR:胰岛素受体;KCNJ:内向整流型钾离子通道亚家族J;UCP:线粒体解偶联蛋白
CHI目前主要分为三大类型:局灶型、弥漫型、嵌合型。手术资料显示局灶型和弥漫型所占比例大致相当。典型的弥漫型CHI病理特征为胰岛B细胞的细胞核增大,不同胰岛细胞之间的细胞核大小不一,高尔基体中的胰岛素原增加。弥漫型CHI多数为钾离子通道基因ABCC8和KCNJ11突变。局灶型表现为胰腺某一区域腺瘤样增生,多数病例肉眼可见增生直径约为2~10mm。
HH的实验室诊断存在争议,目前仅有美国和日本儿科内分泌协会提出了诊治指南。指南一致推荐所有实验室检查均应在低血糖时进行。由于胰岛素的半衰期短(6分钟),实验室测量可能不准确,错过了胰岛素分泌的峰值。因此游离脂肪酸、β-羟丁酸水平有助于诊断。当患者血糖低于3.0mmol/L时,游离脂肪酸< 1.7mmol/L,β-羟丁酸< 1.8mmol/L;血浆胰岛素、C肽可以检测到;胰高血糖素刺激试验阳性(血浆葡萄糖升高大于1.7mmol/L)可考虑诊断为HH。氟-18标记左旋多巴-正电子发射计算机断层扫描(18FDOPAPET)是目前公认的术前定位“金标准”。这一技术的特异性达到100%,敏感性为88%~94%。
治疗的目的是在正常的饮食下使血糖维持在正常范围内。
1.持续喂养
对于高胰岛素血症伴高氨血症患者,需限制蛋白质摄入;静脉滴注葡萄糖,维持血糖在3.0mmol/L以上。
2.药物治疗
二氮嗪是治疗CHI的首选药物。它与ATP敏感性钾通道的磺酰脲类受体1(SURl)亚单位结合,使钾通道处于开放状态,抑制胰岛素的分泌。二氮嗪用量为5~15mg/(kg·d),分2~3次口服。其他药物包括奥曲肽、胰高血糖素、硝苯地平等。
3.手术治疗
对于药物治疗无效者应选择手术治疗,手术前需行18F-DOPA核素扫描确定病灶类型和位置。局灶型CHI可通过切除病灶使CHI得到治愈。弥漫型CHI患者如药物治疗无效,需行胰腺大部切除术(95%~98%)。约1/3弥漫型患者可通过手术治疗改善血糖。随访结果提示在接受胰腺大部切除患者中,47%的患者在10~20岁出现糖尿病。其他来自欧洲的研究显示,胰腺大部切除手术导致91%以上患者发生糖尿病。发生胰腺外分泌功能障碍者约占39%~49%,此类患者应补充胰酶。
关于CHI诊断及治疗的研究正在不断进步,基因诊断已经成为CHI临床诊疗决策中不可或缺的一部分。但全外显子组测序只能找到50%患者的致病基因突变,仍有约50%患者病因不明。推测内含子区和5′非翻译区(5′-UTR)基因突变也与本病有关,但全外显子组测序无法检测到。进一步寻找CHI的致病基因和对胰岛功能的深入研究是未来CHI发病机制研究的一个主要发展方向。另外,在ABCC8基因突变的CHI患者中,仍有50%内科治疗无效,不得不进行胰腺大部切除。多项临床研究着眼于开发新的药物以治疗ATP依赖钾通道相关高胰岛素血症(ATP-sensitive potassium channel hyperinsulinism,KATP-HI),避免胰腺大部切除术带来的副作用。由于ATP敏感性钾通道的活性受到镁离子等其他因素影响,理想的ATP敏感性钾通道开放剂的筛选成为难点。更理想的药物、新的简便的影像学检查方法以及发现更多致病基因是下一步的研究方向。









