


 收藏
收藏 已收藏
已收藏英文名称 :Waardenburg syndrome
中文别名 :Waardenburg综合征;听力-色素综合征
瓦登伯格综合征(Waardenburg syndrome,WS)又称Waardenburg综合征或听力-色素综合征,是一种以感音神经性耳聋及组织器官色素异常改变为主要临床表现的遗传病,临床上少见。1951年由荷兰医师P.J. Waardenburg首次报道了该疾病,故以其名字命名。WS在世界范围内均有病例报道,没有性别和种族间差异。总发病率约为1/50 000~1/42 000,在聋哑人群中发病率为0.9%~2.8%,占先天性耳聋的2%~5%。目前暂无该病在中国人群中发病率的流行病学报道。
在众多学者提出的导致WS的临床特征假说中,以神经嵴发育不全理论,目前最被认可。神经嵴(neural crest,NC)由神经嵴细胞(neural crest cell,NCC)组成,是一类起源于背神经管的多潜能细胞群。在胚胎发育早期沿背神经管迁徙的过程中逐渐分化为黑素细胞、交感神经节的神经元细胞、周围神经与肠道神经系统的神经胶质细胞、颅面骨的骨和软骨细胞以及内分泌细胞等,参与许多组织器官的发育。包括色素系统、肌肉骨骼系统、循环系统、内分泌腺和周围神经系统。当NCC在增殖、生存、迁徙和分化的过程中,任何阶段或空间上发育异常都会导致相应细胞、组织和器官出现异常而致病,统称为神经嵴病。WS即为其中之一,是由于NCC发育异常而导致的一组临床综合征。
研究表明,由神经嵴分化而来的黑素细胞主要表达于全身真皮、表皮、眼睛的脉络膜,而黑素细胞最主要的功能是产生黑素以确保毛发、皮肤和眼睛等组织器官的色素沉着。在内耳发育过程中,黑素细胞迁移至内耳形成中间细胞,与边缘细胞共同发育成血管纹结构,并相互之间形成致密连接。其包含的钾离子通道为耳蜗内电位的产生提供主要能源。因此黑素细胞的存活、增殖和分化缺陷,直接导致WS患者的色素异常和耳聋。同时由于颅面骨、四肢骨骼肌肉以及肠壁神经节均来源于胚胎的NCC,因此也可能会导致这些组织和器官的发育异常,从而产生了WS的一系列伴随症状。
目前较为认可的WS发病机制有两种学说——单倍体剂量不足学说和显性负效应学说,但二者只能解释部分WS发病机制。其中单体倍剂量不足学说认为突变蛋白可能仍有功能,但其剂量不足以实现细胞完全发育,不同个体症状严重程度的差异可能是由于残留的正常野生蛋白量的不同以及不同个体不同器官中细胞发育所需野生蛋白量不同所致,显性负效应学说认为,在两个等位基因中如果一个基因突变,另一个基因保持野生型,即使突变的基因完全失去功能,理论上这一对等位基因仍应保留50%功能。但某些情况下突变的蛋白质不仅自身不能发挥正常生理功能,还使正常蛋白质也不能发挥功能,这种蛋白质相互作用中的干扰现象称为显性负效应。而由于WS致病基因多,且各基因表达产物之间相互作用影响,导致WS的发病机制十分复杂,仍需要大量研究来进一步明确各基因之间的作用机制。
根据WS具有的不同特征性临床表现进行定义分类,WS共可分为四型,而Ⅱ型和Ⅳ型根据致病基因或突变的不同可进一步划分亚型(表1)。临床上以Ⅰ型(图1)及Ⅱ型(图2)最为常见。1992年Farrer等提出了瓦登伯格综合征Ⅰ型的诊断标准(表2):被诊断为瓦登伯格综合征Ⅰ型的患者必须符合两条及以上主要标准,或者符合一条主要标准与两条次要标准。其他三型依据不同的伴随症状或体征,在Ⅰ型基础上做出分型诊断。
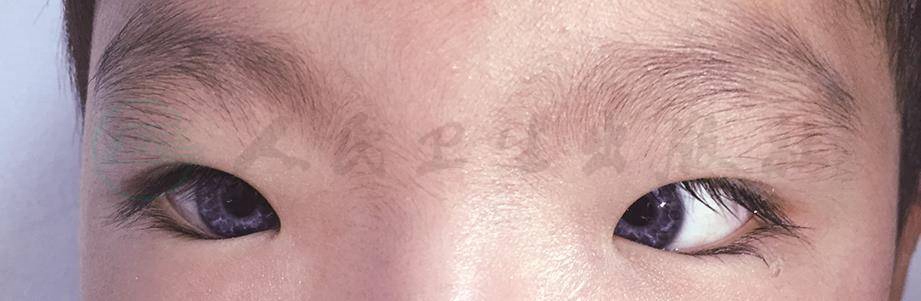
图1 瓦登伯格综合征Ⅰ型
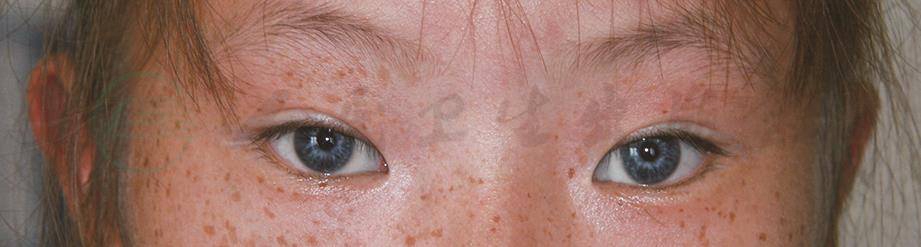
图2 瓦登伯格综合征Ⅱ型
表1 瓦登伯格综合征各型遗传特征
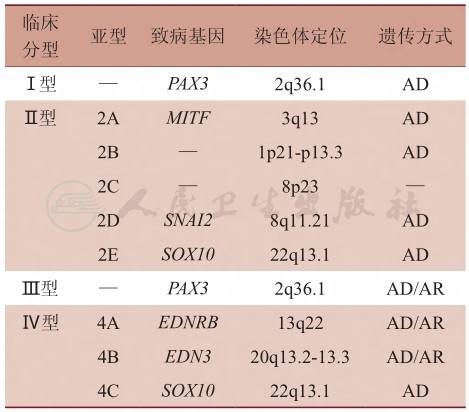
PAX3:配对盒3(paired box 3);MITF:小眼畸形相关转录因子(microphthalmia-associated transcription factor);SNAI2:Snail家族转录抑制因子 2(Snail family transcriptional repressor 2);SOX10:性别决定区盒3(sex determining region Y-box 10);EDNRB:内皮素受体 B(endothelin receptor type B);EDN3:内皮素 3(endothelin 3);AD:常染色体显性遗传;AR:常染色体隐形遗传
表2 瓦登伯格综合征Ⅰ型诊断标准

*W指数的计算:a =内眦间距,b =瞳间距,c =外眦间距,W指数 = X + Y + a/b,X =(2a − 0.2119c − 3.909)/c,Y =(2a − 0.2749b −3.909)/b
瓦登伯格综合征Ⅱ型的诊断标准1995年由Liu等提出,在Ⅰ型的5项主要诊断标准中去除内眦异位,加入早发白发,符合2项及以上主要标准才可被诊断为瓦登伯格综合征Ⅱ型。
瓦登伯格综合征Ⅲ型又称Klein-瓦登伯格综合征,在瓦登伯格综合征Ⅰ型基础上合并上肢肌肉骨骼发育异常。表现为肢体肌肉发育不良或肘部、手指关节挛缩。
瓦登伯格综合征Ⅳ型又称Waardenburg-Shah综合征或Waardenburg-Hirschsprung病,其临床表现与Ⅱ型相似,主要特征是合并有Hirschsprung病,表现为先天性巨结肠或胃肠道闭锁。另外部分Ⅳ型患者可出现包括周围神经病变、智力迟钝、小脑共济失调等神经症状。
WS具有高度的遗传异质性,其遗传方式主要是常染色体显性遗传,部分也可表现为常染色体隐性遗传。已证实有6种基因与该病有关,即MITF、PAX3、SOX10、SNAI2、ENDRB、EDN3,其中PAX3、MITF、SNAI2和SOX10为转录调控因子,EDN3和ENDRB为信号分子(表2)。WS多是由这些基因单独发生突变所致。这些基因之间相互作用以及与其他基因的相互作用构成复杂的调控网络,调控神经嵴源性细胞和组织尤其是黑素细胞的发育,表现为以MITF为中心的调控与被调控的功能性联系。研究显示,MITF在黑素细胞中特异性表达是调节黑素细胞发育的关键转录因子和总调控因子,参与黑素细胞生存、迁徙、增殖和分化的发育过程,可直接调控黑色素生成的三个关键酶的表达,即酪氨酸酶(tyrosinase,TYR)、酪氨酸酶相关蛋白1(tyrosinase related protein 1,TYRP1)和多巴色素互变异构酶(dopachrome tautomerase,DCT)。PAX3与SOX10可单独或二者协同激活并上调MITF表达。MITF对DCT的转录激活需要与SOX10协同作用,而PAX3则拮抗MITF对DCT的转录激活作用。此外,SOX10还能够直接调控EDNRB的表达,而EDNRB又可通过信号传导通路调控MITF转录表达。除了基因突变外,遗传背景、基因修饰、环境、个体差异、起病时间等多种因素都能够影响其临床表型。因此,不同的基因突变在不同家族间、同一基因突变在不同家族或同一家族内不同个体都会产生较大的临床表型差异。
家族中有出现上述相关临床表现的成员,所有同血缘亲属均应接受临床专科检查和基因检测以明确诊断。该病患者的后代应被视为先天性耳聋的高危人群,出生后应行听力全面检查,早期明确诊断,早期干预。已明确诊断的患者孕育下一代前可进行产前咨询,评估下一代患病风险。如已知致病基因,还可进行产前基因检测了解胎儿是否遗传该病,但由于该病的外显不全,无法预测症状的严重程度。
尚无特异的针对病因治疗方法,临床上主要针对患者相关症状进行对症治疗:对于伴有先天性感音神经性耳聋的患者,可尽早行佩戴助听器干预;耳聋程度较重者,可考虑行人工耳蜗植入手术。其他如神经系统病变、肢体畸形及先天性巨结肠等其他症状的治疗主要以对症支持治疗为主,必要时行手术治疗。
随着科学研究手段的不断丰富,国内外研究小组针对WS的遗传突变检测、发病机制等进行了大量研究工作,并取得了较大的研究进展。然而WS致病基因较多,且各致病基因之间相互作用形成网络,机制复杂,至今尚未完全研究清楚。临床上仍有部分WS患者的致病基因没有发现,且WS具有高度的临床异质性,这些因素给该病的分型诊治造成了很大的困难。在治疗方面,人工耳蜗植入对于WS感音神经性聋患者是目前唯一有效的干预手段。但由于国内外对于WS各型人工耳蜗植入术后的疗效尚无全面研究和评估,术后效果仍然存在不确定性。近年来,已有学者探索采取诸如基因打靶、基因转染等基因工程技术尝试实施基因治疗,但均未应用于临床。希望在今后的相关研究中能进一步加强对WS遗传突变鉴定、发病机制、治疗等方面的研究,为临床上早期预防和诊治WS提供理论和实验依据。而随着发病机制的明确以及基因筛查、基因诊断和基因治疗技术的进一步发展和完善,将为更多的WS患者及家庭提供帮助。









