


 收藏
收藏 已收藏
已收藏英文名称 :auditory neuropathy
听神经病(auditory neuropathy,AN)是一种特殊的听觉功能障碍性疾病,它描述了一种内毛细胞、带状突触、螺旋神经节和/或听神经本身功能不良所致的听觉信息处理障碍。临床主要表现为患者可以听到声音却不能理解其语义,患者的听觉时域处理功能下降,言语识别率与纯音听阈不成比例地下降;外毛细胞的功能正常——耳声发射(otoacoustic emissions,OAE)和/或耳蜗微音电位(cochlear microphonic,CM)可引出,而听神经功能异常——听性脑干反应(auditory brainstem response,ABR)异常或全部消失,同时多伴有中枢或周围神经病变。
听神经病是20世纪90年代以来逐渐被认识和发现的疾病。1992年,我国学者顾瑞将其称之为“中枢性低频感音神经性听力减退”。1996年,美国学者Arnold Starr等随访追踪了他们在1991年报道的1例患者(他们称其为EVE,夏娃),同时总结了10例类似临床表现的患者的听力学特征,将此类疾病命名为“听神经病”。听神经病是导致婴幼儿及青少年听力言语交流障碍的常见疾病之一,约1/7 000的新生儿存在听神经功能异常,占儿童永久性听力损失的10%。根据WHO定义的罕见病发病率范畴(0.65‰~1‰),听神经病属于罕见病范畴。
随着听神经病患者的临床遗传学研究和听觉生理学的分析以及动物模型的机制研究进展,导致疾病的相关基因及功能也逐渐明晰。在此,主要针对与听突触病密切相关的OTOF基因(otoferlin)、SLC17A8(VGluT3)基因以及与听神经及周围神经病变相关的OPA1、AIFM1、MPZ、PMP22、PJVK及DIAPH3等基因的发病机制和临床意义简要总结,见表1。
(一)听突触病
由OTOF基因导致的听突触病,OTOF的无义或截短突变导致内毛细胞(inner hair cells,IHCs)中Ca2+触发的胞吐作用几乎停止,错义突变会降低毛细胞中的otoferlin蛋白水平,且囊泡补充的缺陷几乎完全损害了内毛细胞突触的声音编码功能。携带OTOF基因错义突变的患者的心理物理学和生理学测试提示传入信号渐进性减弱,产生听觉疲劳现象。OTOF基因在成人及婴幼儿中的突变频率及临床表型特征以及温度敏感性患者的突变特征研究发现OTOF基因致病变异是婴幼儿听神经病的主要病因之一。
由SLC17A8基因变异也可导致听突触病。缺乏由SLC17A8基因编码的VGluT3蛋白的IHCs表现为毛细胞带状突触中囊泡摄取功能障碍,使囊泡含有的谷氨酸水平降低,进而释放到突触间隙的谷氨酸水平下降,不足以在一级神经元中产生动作电位,没有兴奋性传入突触信号传递到螺旋神经节(spiral ganglion neurons,SGNs),听觉通路的声诱发活动也检测不到,最终导致毛细胞突触信号传递缺陷。在患者听力损失的致病机制方面,听力障碍的发生机制中有两个观点:一个观点是单倍体剂量不足可能会导致出现一个无功能的SLC17A8等位基因;另一个观点是功能增强型突变会导致信号传导增强,这会使内毛细胞突触由于囊泡谷氨酸负荷增加而出现兴奋毒性突触损失。另外,携带SLC17A8基因突变的个体中观察到听力障碍伴有精神和运动发育迟缓,认为与SLC17A8基因突变有关的听力障碍也可能是综合征型的一种表型。
(二)听神经病
由OPA1基因变异导致遗传性听神经病,OPA1蛋白C末端的截短突变主要是由于单倍剂量不足导致非综合征型常染色体显性视神经萎缩(DOA);错义突变可能通过突变蛋白的显性负效应而导致综合征型常染色体显性视神经萎缩(DOA+)。DOA+相关的听力障碍主要由OPA1基因Arg445His错义突变导致,该突变可引起视神经和听神经脱髓鞘及突触丢失。
表1 听突触病和听神经病遗传特征
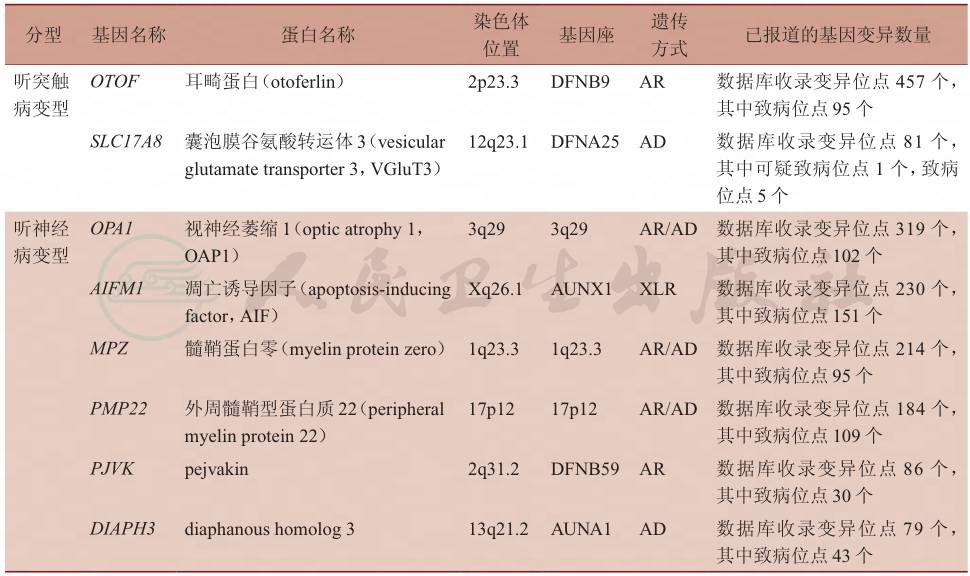
AR:常染色体隐性遗传;AD:常染色体显性遗传;XLR:X连锁隐性遗传;数据库来源:https://www.ncbi.nlm.nih.gov/clinvar/
由AIFM1基因变异导致听神经病,为笔者课题组克隆的致病基因,其结构包括两个黄素腺嘌呤二核苷酸(FAD)结构域、一个还原型烟酰胺腺嘌呤二核苷酸(NADH)结合区和一个具有凋亡前活性的C末端结构域。在N末端还有一个线粒体定位序列(MLS)。在第一个FAD结构域和MLS之间,存在一个潜在的跨膜结构域(TM)。此外,凋亡诱导因子AIF(apoptosis inducing factor,AIF)还含有两个DNA结合位点,以及热激蛋白70(HSP70)、亲环素A(CypA)的结合区。AIF蛋白功能主要是诱导细胞凋亡,参与调控线粒体的结构和氧化代谢过程,影响呼吸功能。AIF是作为caspase-非依赖性凋亡效应分子被发现的,在凋亡损伤时由线粒体转运至细胞核,诱导细胞凋亡。在听觉通路中,无论是内毛细胞,神经通路中的胶质细胞,还是螺旋神经节细胞等,正常的能量代谢是其维持其生理活性的关键。
由MPZ基因导致听神经病的发病机制是MPZ的杂合突变可以通过功能丧失减少正常蛋白的总体数量,或通过显性负性作用破坏MPZ复合体的形成或正常功能。纯合型突变通过功能丧失机制导致MPZ的完全缺失,从而产生周围神经的脱髓鞘病变。听觉神经纤维突触丢失与脱髓鞘通过减少传入及减缓动作电位的传导而导致听觉信号时间编码紊乱。
由PMP22基因导致听神经病是由于PMP22点突变导致严重的髓鞘发育不全或脱髓鞘。变异的PMP22蛋白常常会在内质网或高尔基复合体中形成蛋白质聚积体。这些变异蛋白质聚积体还可能阻断正常PMP22蛋白向细胞膜的运输。
由PJVK基因导致听神经病的病理机制是PJVK基因编码的蛋白pejvakin主要表达于耳蜗Corti器、螺旋神经节细胞以及前三级听觉传入通路(耳蜗核、上橄榄复合体、下丘)的神经元细胞中,所致听神经病的病变部位主要位于听觉传导通路,影响动作电位的传导及细胞内物质交换,而内毛细胞功能不受影响,导致的听神经病以突触后型为主。
由DIAPH3基因导致听神经病的可能机制是既可导致突触前病变也可引起突触后病变。Diaphanous蛋白既存在于果蝇的神经肌肉接头处的突触前成分,也存在于其突触后成分。DIAPH3基因可调控突触前肌动蛋白,维持细胞及静纤毛形状、囊泡转运,调控微管细胞骨架的活动,故DIAPH3基因突变时可导致突触前病变。DIAPH3基因也可上调基因表达,使听神经纤维末梢树突形态发生改变,影响螺旋神经节细胞树突棘的功能,产生迟发性的毛细胞功能损伤,导致突触后病变。
随着对听神经病的听觉生理与遗传学病因的研究进展,可将听神经病分为5种类型。①内毛细胞型:累及内毛细胞本身的突触前病变;②突触型:累及内毛细胞带状突触的突触前病变;③树突型:累及无髓鞘听神经树突的突触后病变;④节细胞型:累及螺旋神经节细胞的突触后病变;⑤轴突型:累及有髓鞘神经轴突的突触后病变。
根据上述累及的病变部位及发病机制,临床诊断可分为三种。
1.听突触病(曾经称之为Ⅱ型听神经病)
涵盖上述①②部位,为突触及突触前型病变,累及耳蜗内毛细胞和/或内毛细胞带状突触,而听神经纤维正常。
2.听神经病(曾经称之为Ⅰ型听神经病)
涵盖上述③④⑤部位,为突触后型的听神经病变。累及无髓鞘听神经树突、螺旋神经节细胞、有髓鞘神经轴突、髓鞘等听神经纤维,而内毛细胞及其突触正常。其中明确由遗传基因导致的听神经病变称为遗传性听神经病。
3.非特异性听神经病
突触前后均受累的病变称为非特异性听神经病。
一、临床干预
(一)改善信噪比
理论上,任何能够提高信噪比的方法都可以提高听神经病患儿的言语识别和语言学习能力,所以在结构性或自然语言学习过程中,可以通过减少环境噪声、利用扩音器增加说话者音量、使用FM系统等方法实现信噪比的优化,帮助婴幼儿听神经病患者改善交流能力,但实际效果较为有限。
(二)验配助听器
临床上听神经病患者的干预手段主要以助听器和人工耳蜗植入为主。但助听器对听神经病患者的康复效果存在个体差异,是否能使患者最终受益尚有争论。尽管有些学者认为儿童听神经病患者佩戴助听器的受益与耳蜗植入后平均受益水平相当,但目前仍然缺乏更有力的证据。
(三)人工耳蜗植入
人工耳蜗植入治疗听神经病已有较长时间,听神经病患者的耳蜗植入效果具有多样性。有些学者认为,耳蜗植入可使大部分语前聋的听神经病患儿言语识别能力有显著提高,但也有一些学者指出部分(约25%)听神经病患者耳蜗植入术后效果不佳,未获得有效的听力改善。耳蜗植入的效果与病变部位密切相关。根据现有研究,突触前听神经病患者显示出和普通感音神经性聋相似的术后获益。突触后病变的患者手术效果各异,但平均差于突触前患者的术后效果。另外,听神经病患者耳蜗植入前详细的MRI和CT检查非常必要,以确定患儿是否存在听神经发育不良和耳蜗神经缺如的情况,从而判断术后的效果,研究表明仅有不到10%的蜗神经缺如病例可通过人工耳蜗植入获得言语感知能力改善。
(四)药物治疗
除了助听器和人工耳蜗植入,目前关于药物治疗听神经病仍处于探索阶段。婴幼儿听神经病和青少年的发病类型和机制不同,针对听突触功能恢复和修复听神经髓鞘的药物,以及未来的基因治疗将成为一种可期待的解决方案。
二、未来展望
基因治疗是未来对特定类型听神经病的可行的治疗方式。Akil等通过基因治疗使小鼠成功恢复听觉,这是耳聋基因治疗方面的重大进展。Wan等研究表明,编码耳蜗类神经胶质支持细胞中神经营养因子3(neurotrophin 3,Ntf3)基因的过表达促进噪声引起损伤后突触数目和功能的恢复。Cunningham等也进一步验证了噪声引起的突触病变是可恢复的,甚至提高了其在暴露于破坏性噪声后进行治疗的可能性。虽然这些研究结果很乐观,但是还需要更多的时间才能将这些治疗方法应用于听神经病患者。而我们在临床上发现的OTOF基因突变患者(先天性)以及AIFM1基因突变患者(迟发型),后者拥有更好的时间窗来进行未来基因治疗的研究与尝试,具有可期待的治疗前景。
听神经病从发现到渐入精准经历了20多年,取得了20年前无法想象的进展,但仍然还有诸多机制有待挖掘和发现,对听神经病的毛细胞声音编码与突触功能的研究以及未来的基因治疗研究仍然是今后的主要方向;推广基于精准医学的分子分型定位诊断以及个性化康复干预是目前有效的方法与途径。
(一)一级预防
目的在于预防疾病的发生,建议开展聋病基因检查及咨询。产前诊断、羊水检查等以预防相关AN患儿的出生,防范氨基糖苷类抗生素的中毒等。
(二)二级预防
目的主要消除引起AN的病因,做好产前和围产期保健(及时处理新生儿高胆红素血症、核黄疸及围产期窒息、缺氧等);及时处理神经脱髓鞘病变、炎性神经病变、缺血/缺氧性神经病、脑积水、大脑性麻痹、传染性疾病(流行性腮腺炎)、自身免疫性疾病等。
(三)三级预防
目的在于早期诊断并早期给予康复治疗。①对高危新生儿进行筛查、随访;②对学龄前儿童定期进行听力检查;③如果有条件应用基因芯片筛查,对疑诊AN的患者进行已知AN基因筛查;④AN确诊之后,采取综合康复治疗措施,以提高听力言语水平、社会适应能力,以帮助患者克服各种困难,使患者能达到本人的最佳功能状态。









