


 收藏
收藏 已收藏
已收藏遗传性眼病是一组临床表现多样且危害严重的致盲性眼病,具有高度临床和遗传学异质性。遗传性眼病种类繁多,病程及临床表现各有特征,可以累及眼部的各结构,如眼睑、角膜、晶体、虹膜、房角、玻璃体、视网膜、视神经等。近20年来分子遗传学技术发展迅速,已经发现数百个基因与遗传性眼病相关。由于遗传性眼病大多只致盲不致死,并且容易识别,因而并不十分罕见,在各器官单基因遗传病患病率排第一;在致残遗传病方面,遗传性眼盲仅次于遗传性耳聋,排第二。
以遗传性视网膜疾病为例,不同疾病临床表现差别明显。①视网膜受累细胞先后及轻重不同:视网膜色素变性(retinitis pigmentosa,RP)(OMIM#268000),视杆细胞首先受损且重,逐渐累及视锥细胞;而锥细胞或锥杆细胞营养不良(cone/cone rod dystrophy,COD/CORD)(OMIM #120970)则以视锥细胞首先受损,视杆细胞受累较晚。②受累部位不同:卵黄样黄斑营养不良(vitelliform macular dystrophy,VMD)(OMIM #153700)和Stargardt病(OMIM #248200)主要累及黄斑,而RP首先累及中周部视网膜。③起病时间差异大:Leber先天性黑矇(Leber congenital amaurosis,LCA)(OMIM #204000)出生后即表现出明显视力低下,而多数RP在成年后中心视力才受损。④疾病进程不同:先天性静止性夜盲(congenital stationary night blindness,CSNB)与全色盲(achromatopsia)病情稳定,而大多RP、CORD及黄斑变性病情则进行性发展。还有一些其他遗传性视网膜疾病以综合征的形式表现,如Usher综合征表现为RP合并感音神经性耳聋;Alström综合征表现为CORD合并感音神经性耳聋、肥胖、胰岛素抵抗和肝肾功能异常,Bardet-Biedl综合征表现为RP合并肥胖、多指(趾)畸形、性腺发育不良和肾功能异常,通常都会严重影响患者视力。
遗传性眼病的复杂之处还表现在,不同疾病的致病基因和临床表现有交叉和重叠。一方面,同一种疾病临床严重程度及眼底表现差异很大,如已有20多种基因发现与LCA相关,不同致病基因导致的眼底差异非常明显(图1)。另一方面,同一个致病基因不同的突变可对应几种有显著临床区别的视网膜变性,如鸟苷酸环化酶2D(GUCY2D)基因(OMIM#600179)不同的突变既可以导致常染色体隐性遗传(LCA),也可导致常染色体显性遗传(CORD)(图2)。低密度脂蛋白受体相关蛋白5(LRP5)基因(OMIM #603506)所致临床表现差异最大,可以是典型家族性渗出性玻璃体视网膜病变,也可以表现为斜视、弱视、屈光参差、高度近视、青少年视网膜脱离等。一些常见眼科疾病,其中相当一部分是由单基因突变所致,如早发高度近视、原发性开角型青光眼、视网膜脱离等。遗传性眼病是眼科难治性致盲眼病的重要原因。
目前多种测序方法,如Sanger测序、二代测序、三代测序、基因芯片、MLPA、array CGH等,使人们能够更高效、更全面地检测基因突变情况,提高了基因诊断效率和准确性。致病基因的确定不仅能够有助于解释这类病的发病机制,同时也给遗传性眼病的治疗提供了新的思路。眼球结构相对封闭,基因载体可以只转染到眼特定的靶细胞,视网膜下腔具有高度的免疫赦免性,是基因载体注射的理想部位,这提示遗传性视网膜疾病是基因治疗的理想对象。正在进行的多项临床试验也证明了基因治疗的安全性与有效性。另外干细胞以及新药研发等领域也有可能给遗传性眼病的治疗带来曙光。但就目前水平而言,我国在遗传性眼病的研究进展方面与国外还存在一定差距。因此,我们应该重视此种疾病的基础和临床研究,为遗传性眼病患者带来更多的光明和希望。
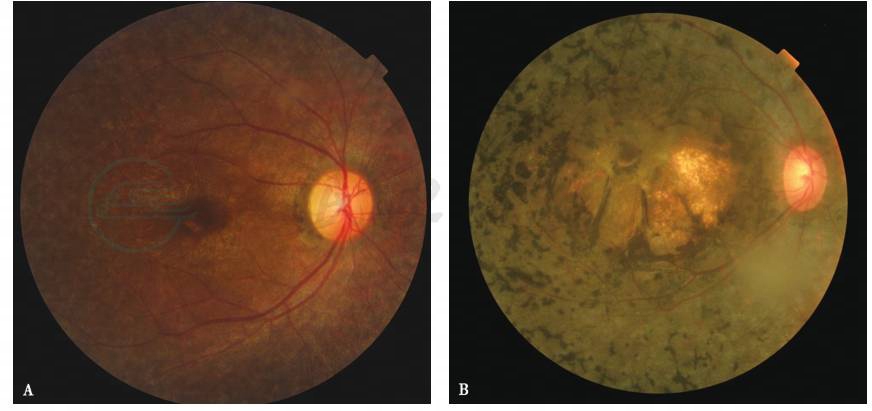
图1 LCA患者不同的眼底表现
A.RPE65突变所致LCA;B.RDH12突变所致LCA
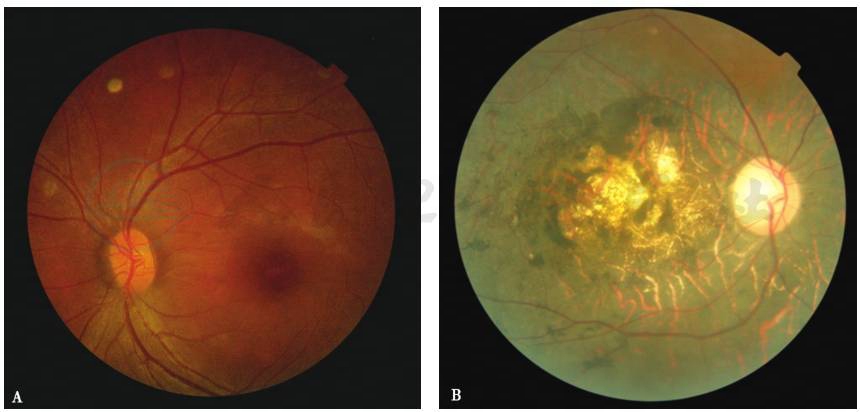
图2 GUCY2D基因不同的突变导致不同的临床表现
A.GUCY2D突变所致LCA;B.GUCY2D突变所致CORD









