


 收藏
收藏 已收藏
已收藏英文名称 :severe combined immunodeficiency disease
联合免疫缺陷病(combined immunodeficiency disease, CID)是一组以T/B细胞缺陷为主,同时伴有不同程度其他细胞缺陷的异质性疾病。目前发现至少16种不同基因突变可导致该病。国外流行病学研究显示,CID患病率约为1/10万~2/10万活产婴。CID中最为严重的类型为重度联合免疫缺陷病(severe combined immunodeficiency disease, SCID),常常引起T细胞数量显著降低,B细胞和NK细胞不同程度降低。临床常表现为生后2~5个月内出现生长发育停滞、持续性腹泻、明显细菌感染、鹅口疮、肺孢子菌肺炎和播散性卡介苗感染等。如不经早期诊断、严格隔离、防治感染、造血干细胞移植或基因治疗,绝大部分患者于2岁内死亡。在引起SCID的16种疾病中,以白细胞介素2受体γ链(interleukin-2 receptor common gamma chain, IL2RG)缺陷所致X连锁重度联合免疫缺陷病最为常见,约占所有SCID的50%,因此本节主要阐述该病。
X连锁重度联合免疫缺陷病(X-linked severe combined immunodeficiency disease, X-SCID)是由IL2RG基因突变引起的细胞免疫和体液免疫联合缺陷。发病率约1/60 000~1/50 000活产婴。IL2RG基因位于Xq13.1,其编码的蛋白γc是IL-2、IL-4、IL-7、IL-9、IL-15和IL-21等细胞因子受体的共同组分。IL2RG基因含4 500个核苷酸,由8个外显子组成,编码389个氨基酸构成的γc蛋白。γc蛋白组成性表达于T、B、NK和髓红系祖细胞表面。IL-2与T细胞的发育和活化有关,IL-4与B细胞的类别转换和Th2细胞的分化密切相关,IL-7与早期淋巴细胞系的发育有关,IL-15和IL-21与NK细胞的发育有关。一旦γc与上述细胞因子结合,将通过下游的Janus激酶3-信号转导和转录激活因子5(JAK3-STAT5)向细胞核传递活化信号,从而改变相关基因转录程序,控制免疫细胞的发育、活化和功能发挥。因此IL2RG基因突变将导致T细胞和NK细胞发育障碍、B细胞功能障碍等免疫异常,典型的X-SCID表现为T−B+NK−的表型。
(一)淋巴细胞计数
绝大部分患儿外周血淋巴细胞减少,绝对计数常常<2.5×109/L,甚至<1.5×109/L。如发生母体淋巴细胞植入,外周血淋巴细胞水平可正常。
(二)淋巴细胞分类
典型X-SCID患者T细胞、NK细胞数量比例显著降低,B细胞数量正常,比例显著上升,但存在功能异常,呈经典T−B+NK−的免疫表型。X-SCID患儿的B细胞是未成熟B细胞,缺乏高频突变,产生免疫球蛋白功能低下。正如上述,部分X-SCID也呈现非经典免疫学表型,这与基因突变类型和母体细胞植入等均相关。
(三)免疫球蛋白
免疫球蛋白常全面低下,由于母源性免疫球蛋白的存在,出生时IgG可正常,3月龄后逐渐下降。需注意,进行血清IgG水平评估时,应除外丙种球蛋白输注的影响。
(四)细胞/体液免疫功能
T细胞对植物凝集素(phytohemagglutinin, PHA)等丝裂原或抗CD3抗体增殖反应异常提示细胞免疫缺陷。疫苗和感染原的特异性抗体反应严重受损或缺乏提示体液免疫缺陷。
(五)T细胞受体重排删除环
T细胞受体重排删除环(T cell receptor rearrangement excision circle, TREC)是T细胞在胸腺发育过程中形成的DNA环,反映T细胞的胸腺输出功能,SCID患儿TREC显著降低,通过定量PCR的方法进行TREC检测,可早期发现SCID患儿。该手段已经用于新生儿筛查,敏感性高。
(六)母源性细胞植入检测
X-SCID患儿常常存在母源性淋巴细胞植入,对诊断有较大指示意义。可通过HLA分型、DNA多态性标记检测XX核型,确定母源性细胞植入。如果采用敏感的方法,几乎所有的X-SCID患儿均可检测到母体细胞。
(七)γc基因mRNA及蛋白表达
γc基因mRNA及蛋白表达可显著降低,但部分患儿基因发生错义突变时,其mRNA表达无变化。外显子7和外显子8突变引起蛋白胞内段异常时,针对胞外段检测抗体可能仍正常结合γc蛋白而呈现正常表达,因此蛋白阳性并不能完全除外诊断,需要进行基因分析。
(八)IL2RG基因分析
IL2RG基因突变是X-SCID确诊依据。各外显子均有突变报道,外显子5最多,突变类型包括错义突变、无义突变、插入突变、缺失突变和剪切位点突变等,其中c.690C>T、c.691G> A、c.684C>T、c.879C>T和c.868G>A等突变频率最高。
(九)其他
胸腺是T细胞发育成熟场所,X-SCID患者常常表现胸腺明显缩小及淋巴细胞缺如,淋巴结和扁桃体亦发育不良。病理检查提示胸腺基质存在分化不良,胸腺树突状细胞及上皮细胞异常。胸部CT、X线检查可发现胸腺影减小或缺如。
X-SCID为儿科急症,一旦确诊,应迅速完成对患儿的评估,包括详细病史、生长发育状况、感染情况等。同时患者宜严格隔离,积极予以IVIG替代治疗、复方磺胺甲噁唑预防肺孢子菌感染、强有力抗生素清除感染,尤其注意真菌、结核、EB病毒、巨细胞病毒、肺孢子菌、原虫的筛查和治疗。禁止接种一切减毒活疫苗,输注血液制品应经过辐照清除具有增殖能力的细胞。尽量延长患者寿命,保护脏器功能,尽可能为移植做准备。
本病唯一根治方法为造血干细胞移植(hemapoietic stem cell transplantation, HSCT)。1968年首例骨髓移植成功,并成为标准的免疫重建手段。宜采用同胞等遗传背景完全相同的供者,尽管部分患儿B细胞重建不理想,HSCT成功率可高达90%以上。X-SCID进行HSCT通常并不需要清髓预处理,有时可完全不用免疫抑制药物,移植后虽然可能仅为嵌合状态,但亦可保全患儿生命。
基因治疗越来越受到关注。X-SCID基因治疗的优势在于不需要寻找HLA配型相合供者,也可避免GVHD的发生。2010年以后,欧美国家进行了X-SCID基因治疗多组临床试验,总体取得了良好治疗效果,由于采用了自灭活病毒载体,新一代基因治疗也尚未发现致瘤副反应。
诊疗路径详见图1。
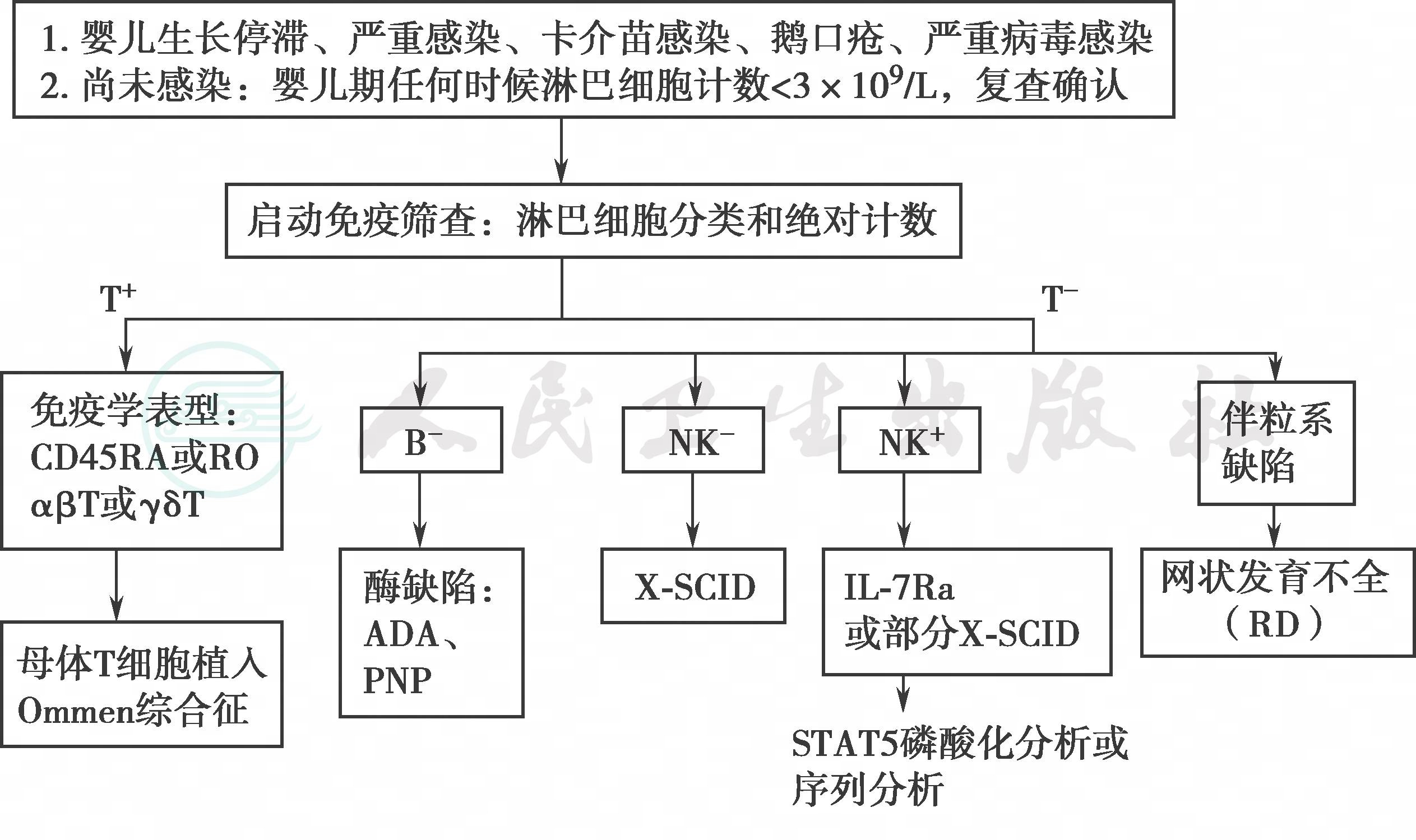
图1 重度联合免疫缺陷病(SCID)诊疗路径
ADA:腺苷脱氨酶;PNP:嘌呤核苷磷酸化酶









