


 收藏
收藏 已收藏
已收藏英文名称 :congenital adrenal hypoplasia
先天性肾上腺发育不良(congenital adrenal hypoplasia,CAH)是一种罕见的疾病,可以通过X连锁(OMIM #300200)或常染色体隐性遗传(OMIM #240200)方式遗传,前者为最常见的病因。大多数受累的患儿在新生儿期可能会因为肾上腺危象危及生命,表现为失盐、低血糖所致惊厥和皮肤色素沉着。盐皮质激素和糖皮质激素的水平明显下降,并且对促肾上腺皮质激素(adrenocorticotropic hormone,ACTH)刺激缺乏反应。除肾上腺皮质功能不全外,低促性腺激素型性腺功能减退症(hypogonadotropic hypogonadism,HH)是X连锁CAH的常见特征,表现为青春期延迟并需要进行睾酮替代治疗。
自从1855年英国的Thomas Addison医生描述了以皮肤色素沉着、乏力、低血压、低钠血症为主要表现的原发性肾上腺皮质功能不全后,此后将这类疾病命名为“艾迪生病(Addison's disease,Addison病)”,特指因各种病因导致肾上腺皮质不能合成足够的皮质醇,ACTH反馈性升高的疾病。1930年左右,Crosby EH等报道了一些成人Addison病的病例,尸检结果找不到肾上腺,当时这些病例被认为是肾上腺未发育,但病因不详。而第1例明确的CAH病例报道来自1948年捷克Sikl H医生的研究,他报道了1例出生33天的男婴,出生时一切正常,但逐渐出现喂养困难、体重不增,就诊时有明显的皮肤色素沉着,患儿出现腹泻、肺炎等严重合并症去世。之后尸检结果提示患儿的肾上腺体积明显缩小,此后文献中出现了越来越多的CAH的病例报道。
CAH有广义和狭义两种概念,广义CAH的发病机制涉及垂体发育异常、ACTH受体抵抗、原发性肾上腺发育缺陷、染色体异常和一些罕见的综合征等(表1),分子发病机制涉及广泛的细胞和生理层面的过程,包括代谢、核蛋白转运、氧化应激防御机制和细胞周期调控。
表1 先天性肾上腺发育不良的广义病因分类
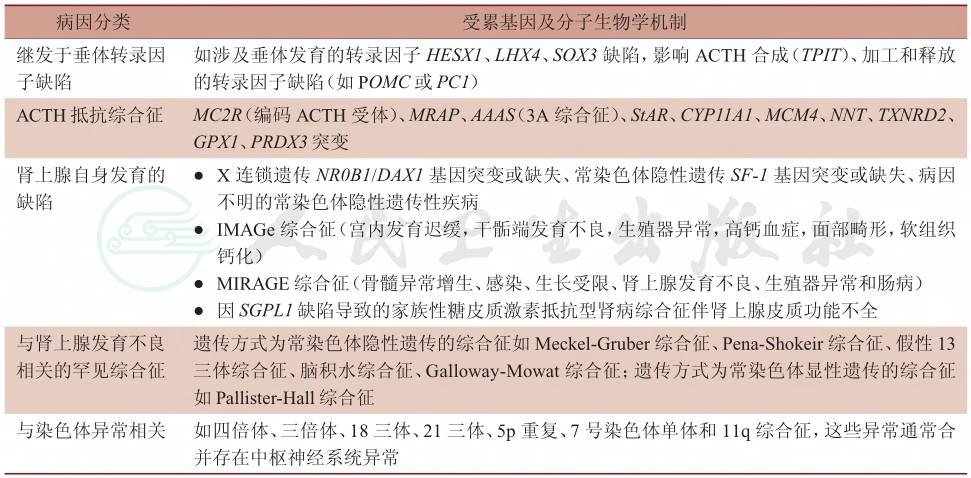
SOX3:Y染色体性别决定区盒基因;ACTH:促肾上腺皮质激素;POMC:阿黑皮素原;MC2R:黑素皮质素受体-2;StAR:类固醇激素合成急性调控蛋白;CYP11A1:细胞色素P450家族11亚家族A成员1;NR0B1/DAX1:核受体0亚家族B组成员1/位于X染色体上剂量敏感的性别反转-先天性肾上腺发育不良关键区域基因1
狭义CAH目前专指由X染色体短臂(Xp21)上的核受体亚家族0 B组成员1(nuclear receptor subfamily 0 group B member 1,NR0B1)基因突变所致,该基因更常用的名称为位于X染色体上剂量敏感的性别反转-先天性肾上腺发育不良关键区域基因1(dosage-sensitive sex reversal,adrenal hypoplasia congenita critical region on the X-chromosome,gene 1,DAX1)。狭义CAH也称为X连锁的CAH(X-linked CAH,XLA)。NR0B1/DAX1基因由2个外显子及1个内含子组成,编码蛋白为核受体超家族的一种孤儿蛋白DAX1蛋白,它在肾上腺皮质、性腺(睾丸间质细胞和支持细胞,卵巢的卵泡膜细胞和颗粒细胞)、下丘脑和垂体均有表达,在肾上腺和性腺的发育中起到了至关重要的作用。因此CAH的典型表现为原发性肾上腺皮质功能不全和HH。
核受体家族在类固醇生成组织的建立和维持中起重要作用,是调节基因网络的转录因子,对生殖、发育和细胞内外信号转导起重要作用。DAX1蛋白羧基端结构域与其他核受体的配体结合结构域具有高度相似性,而DAX1蛋白的氨基端区域是一个不典型的DNA结合域,由66~67个氨基酸序列重复3.5次组成。因此,DAX1蛋白缺乏已知的配体,被命名为孤儿核受体。许多研究表明,DAX1作为受其他核受体调节的类固醇生物合成途径的转录抑制因子来发挥作用,如抑制类固醇生成因子(SF1)介导的StAR基因、3β-羟类固醇脱氢酶和胆固醇侧链裂解酶(P450scc)的转录激活途径;还作为其他核受体如雌激素受体、孕激素受体、糖皮质激素受体和雄激素受体等的抑制物;此外,DAX1蛋白还被认为是一种与细胞核中的核仁蛋白和细胞质中的多核糖体相关的RNA结合蛋白的转运蛋白,提示其在转录后过程中可能发挥额外调节作用。综合以上作用,DAX1在器官发育过程中抑制肾上腺干细胞的分化,从而在祖细胞分化为成熟的类固醇激素细胞之前,扩大祖细胞池。而DAX1功能丧失会导致祖细胞过早分化为成熟细胞,无法维持祖细胞池,从而导致在短暂的肾上腺功能过度活跃后出现肾上腺发育不良。
原发性肾上腺皮质功能不全的发生是因为受累胎儿肾上腺皮质除了胎儿带(fetal zone)以外缺乏皮质永久带(permanent zone),而正常生理状态下胎儿带在出生后逐渐萎缩,而永久带会持续存在,并发育为成人的肾上腺皮质。残余的胎儿带细胞空泡变、体积变大,组织学上被称为“巨细胞”。
本节重点讨论的就是狭义CAH,也就是XLA。CAH为罕见疾病,关于其发病率缺乏相关数据,早期日本的研究推测发病率为1/12 500活产婴儿,而近期的研究估测发病率可能为1/70 000活产男婴。
(一)实验室检查
1.生化检查
可以表现为低钠血症、高钾血症、代谢性酸中毒、低血糖等。
2.肾上腺激素改变
低醛固酮、高肾素水平提示低钠的原因是盐皮质激素的缺乏,17-羟孕酮浓度正常或降低。生命早期可能出现11-脱氧类固醇激素水平升高,如果出现这个现象需要和11β-羟化酶缺陷症鉴别。出生第一周的新生儿患者血清皮质醇浓度变异较大,可以从很低到很高,但年龄更大的婴儿或幼儿发病时血皮质醇水平明显降低,ACTH水平明显升高,ACTH兴奋试验可用于检测是否缺乏皮质醇。
3.性腺激素改变
大多数青春期男性患者的睾酮水平降低,同时促性腺激素水平降低,为HH。促性腺激素释放激素(GnRH)兴奋试验反映垂体促性腺激素的储备,最有可能显示促性腺激素分泌受损。人绒毛膜促性腺激素(HCG)兴奋试验评价睾丸产生睾酮的储备能力。但在病程中,也有可能在婴幼儿期出现类似于中枢性性早熟的表现,表现为睾酮水平明显升高伴随促性腺激素水平也升高,原因如上文所述,此时需要与先天性肾上腺皮质增生症鉴别。
(二)影像学检查
肾上腺CT或MRI检查通常显示肾上腺体积小,但也有患者的肾上腺体积正常或存在异位肾上腺。
(三)基因诊断
在排除了21-羟化酶缺乏、肾上腺脑白质营养不良和自身免疫性疾病以外,在原发性肾上腺皮质功能不全的男性患者中,58%的患者可以检测到DAX1突变。1994年Muscatelli F等鉴定出XLA的致病基因NR0B1,之后学者们鉴定出数量众多的突变,目前有超过200种基因突变,三分之二为点突变,包括缺失、剪接位点的突变、错义突变、无义突变和移码突变等。错义突变约占XLA的四分之一,大多数错义突变倾向于聚集在DAX1的羧基端,表明配体结合域对DAX1蛋白生物学功能的重要作用。大片段缺失通常累及相邻的基因缺失,包括甘油激酶和进行性假肥大性肌营养不良基因,这些患者同时伴随智力发育迟缓。多重连接探针扩增(multiplex ligationdependent probe amplification,MLPA)分析是检测CAH患者NR0B1和相邻基因缺失的一种有价值的工具,MLPA具有识别女性携带者的优势。
临床疑诊肾上腺皮质功能不全的患儿,首先需明确是否为XLA。
需要进行的初步检查如下:①电解质、血糖;②清晨血ACTH、血皮质醇;③血浆肾素活性、醛固酮;④17-羟孕酮;⑤卵泡刺激素(FSH)、黄体生成素(LH)、睾酮;⑥染色体核型;⑦肾上腺影像学(CT或MRI)检查。
如果诊断存在疑问,可以通过以下功能试验来评价:①ACTH兴奋试验评价是否存在肾上腺皮质功能不全;②GnRH兴奋试验评价垂体促性腺激素的储备;③HCG兴奋试验评价睾丸产生睾酮的储备能力。
尽可能进行基因检测来明确诊断。
(一)肾上腺皮质功能不全的治疗
XLA导致肾上腺皮质功能不全与其他病因相似,大多数患者需要同时替代糖皮质激素(氢化可的松)和盐皮质激素(氟氢可的松)。氢化可的松常规替代剂量为10~15mg/(m2·d),分3次服用。在肾上腺危象时,氢化可的松按照(50~75mg/m2或1~2mg/kg)作为初始剂量静脉注射,之后按照50~75mg/(m2·d),每天分4次静脉注射。
(二)性腺功能减退的治疗
XLA的性腺功能减退多数为HH,可以采取睾酮替代疗法来保持男性第二性征。睾酮制剂有口服、肌内注射、贴皮剂和凝胶等多种剂型。也可采用HCG和人绝经期促性腺激素(HMG)来进行生精治疗,帮助患者获得生育能力。但HCG+HMG的双促治疗效果个体化差异较大,部分患者对双促治疗无反应。
(三)临床诊治要点
1.对于孤立性肾上腺皮质功能不全的男性婴幼儿,需要筛查XLA。
2.典型症状为男性原发性肾上腺皮质功能不全和HH;女性携带者通常无症状,可能有青春发育延迟,极个别女性有上述类似症状。
3.XLA性腺激素异常表现多样,典型表现为HH,多于青春发育期被发现,但在青春发育前也可以有中枢性性早熟、周围性性早熟,容易误诊为先天性肾上腺皮质增生症中的21-羟化酶缺陷症。
4.激素检查特点为皮质醇低,ACTH水平明显升高,电解质可能有低钠和高钾;睾酮水平降低,促性腺激素水平低。
5.基因检测是确诊的金指标。









