


 收藏
收藏 已收藏
已收藏肾上腺皮质与卵巢有许多相似之处:①在胚胎发育过程中,两者均由泌尿生殖嵴上皮发育而来;②两者均能合成甾体激素,有共同的初始前身物——胆固醇或乙酸盐,肾上腺皮质网状带也产生甾体性激素;③控制肾上腺与卵巢功能的下丘脑激素释放激素间产生交叉作用,肾上腺皮质与卵巢功能密切相关,肾上腺疾病可以影响卵巢功能,导致月经紊乱和闭经。
1.肾上腺皮质功能亢进
1912年Harvev Cushing描述了一个23岁女性出现肥胖、多毛和闭经,20年后证实该患者有垂体嗜碱性细胞腺瘤;之后将各种原因,包括促肾上腺皮质激素(adrenocorticotrophic hormone,ACTH)过多或肾上腺肿瘤所致的肾上腺皮质功能亢进,表现为向心性肥胖、高血压、高血糖、皮质醇增多、多毛、痤疮、月经失调、不育等一系列症状称为库欣综合征(Cushing’s syndrome),其临床特点见表1。
表1Cushing综合征的临床症状与体征
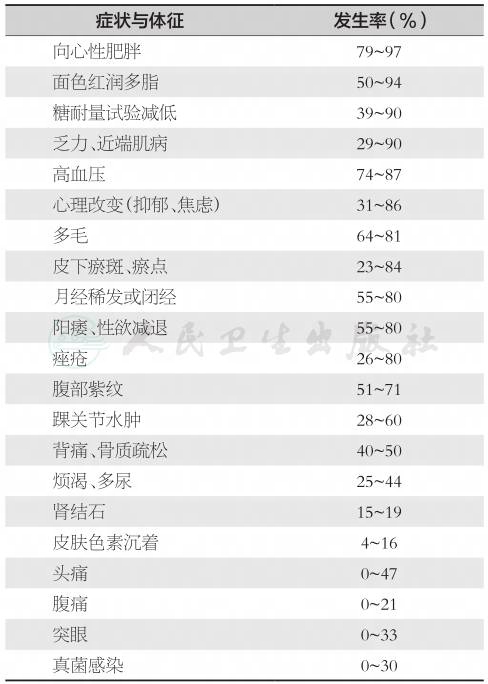
库欣综合征患者月经失调的机制尚未完全阐明,可能包括中枢和外周因素。外周机制指血睾酮水平升高和SHBG降低,脂肪组织增多,使肾上腺来源的雄激素在外周转化为雌酮增多,雌酮过多又干扰下丘脑-垂体-卵巢轴功能。中枢机制指库欣综合征患者闭经可能与下丘脑激素促皮质释放因子(corticotrophin releasing factor,CRF)及5-羟色胺过度分泌抑制GnRH脉冲分泌有关。肾上腺或卵巢分泌雄激素的肿瘤极为罕见,但应考虑可能发生在快速发育不良或血清雄激素显著升高的患者。如果存在皮质醇过量的躯体症状,可通过24小时尿游离皮质醇、深夜唾液皮质醇或地塞米松抑制试验排除库欣综合征。
有报道原发性色素性结节性肾上腺皮质疾病(primary pigmentednodal adrenocorticoid disease,PPNAD)是ACTH非依赖性库欣综合征的一个非常罕见的病因,特别是当肾上腺在影像学上似乎正常,在大剂量地塞米松试验中观察到皮质醇水平的矛盾升高时。患者高皮质醇血症可导致月经周期紊乱,甚至继发闭经、不孕、多毛和痤疮。在绝大多数患者中,PPNAD与卡尼复合体(Carney complex,CC)有关,因此,这些患者及其一级亲属应时刻仔细筛查PPNAD、CC症状,以及PRKAR1A、PDE11A和PDE8B基因的基因突变。
2.肾上腺皮质功能低下
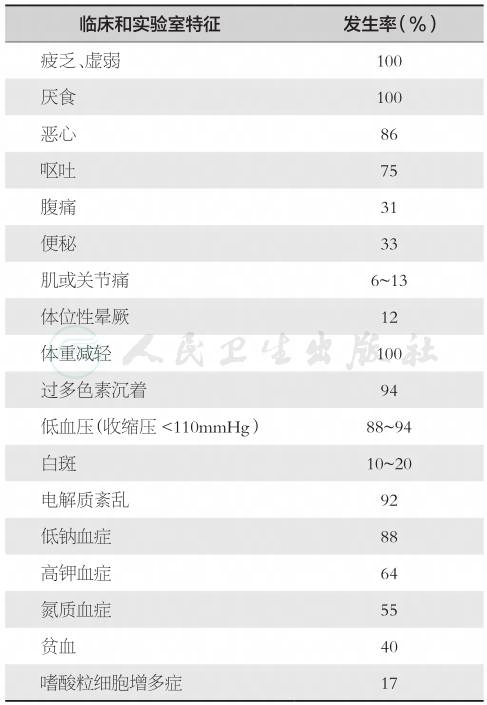
1855年,英国医生Thomass Addison发现一种奇怪的病,临床表现为虚弱、极度疲乏、心动微弱、恶心、呕吐、腹泻、皮肤呈青铜色。通过尸体解剖发现患者双侧肾上腺有破坏性病变,肾上腺皮质功能低下,为原发性肾上腺皮质功能低下,命名为Addison病。Addison病可由肾上腺结核所致,或因肾上腺梅毒、肿瘤、出血等引起,约占全部病例的20%,也可由于长期精神过度紧张,使下丘脑-垂体-肾上腺系统过度兴奋,最后趋于衰竭,肾上腺萎缩。近年来发现自身免疫性疾病是导致Addison病的主要原因,约占75%,可同时合并卵巢、甲状腺、甲状旁腺等自身免疫性疾病,构成多腺体自身免疫综合征(multiple endocrine deficiency syndrome),即Schmitt综合征。肾上腺皮质功能低下时,下丘脑-垂体失去对肾上腺皮质激素的负反馈调节,CRF和ACTH升高,两者均可干扰性腺轴功能。肾上腺皮质功能低下,患者恶心、呕吐、腹泻、体重减轻、低钠血症、失水、低血压、低血糖、无力、晕厥、皮肤色素沉着,营养不良;由于多是自身免疫性疾病引起的多器官功能低下在肾上腺的表现,约22%免疫异常性肾上腺功能低下者可检测出抗卵巢抗体,20%患者出现卵巢功能衰竭,常出现卵巢功能低下,表现为阴毛腋毛稀疏、脱落、性欲减退,病程初期或轻症患者月经变化不大,晚期或重症患者出现排卵障碍、月经增多、闭经、不孕(表2)。
表2肾上腺皮质功能低下的临床和实验室特征
3.先天性肾上腺皮质增生症
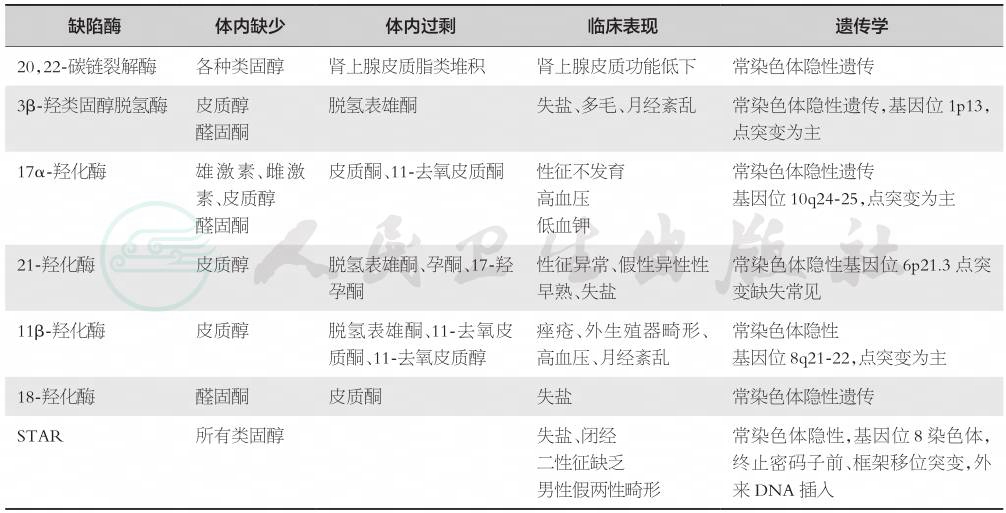
皮质激素合成主要有6种酶,即20,22-碳链裂解酶、3β-羟类固醇脱氢酶、17α-羟化酶、21-羟化酶、11β-羟化酶和18-羟化酶,任何一种酶缺乏,将导致皮质激素缺乏,下丘脑-垂体失去负反馈作用调节、CRF和ACTH产生增加,可刺激肾上腺皮质增生;除外类固醇急性调节蛋白(steroid acute regulatory protein,STAR)基因缺失、突变和异常、外源性DNA插入等引起先天性肾上腺皮质功能低下、18-羟化酶缺乏引起失盐症状外,其余四种酶的缺乏均出现性发育异常或原发性闭经(表3)。晚发型先天性肾上腺增生(如21-羟化酶缺乏)是高雄激素性闭经的常见原因,血清17-羟孕酮水平升高后应行促肾上腺皮质激素刺激试验。
表3肾上腺皮质激素合成酶缺乏的病理生理与临床特征
导致先天性肾上腺皮质增生的酶缺陷中,21-羟化酶缺陷约占患者总数的95%,人群发生率约为0.07‰~0.5‰,阿拉斯加的因纽特人发生率约为1.43‰~3.35‰。该酶是肾上腺皮质醛固酮和皮质醇合成的关键酶,它使孕酮转变为11-脱氧皮质酮,17α-羟孕酮转化为11-脱氧皮质醇。编码该酶的基因位于人类第6号染色体短臂,靠近MHC,由A、B两部分组成,A约长3.2kb,B约为3.7kb。现已发现该酶缺陷主要是点突变所致(E1:Pro30Leu,E4:Ile172Asn,E7:Val281Leu,E8:Arg339His,Arg356Trp,E10:Pro453Ser)和第2内含子的点突变(A/C656G)导致酶活性降低而表现为失盐和/或性征异常,部分位点突变(E6:Ile236Asn,Val237Glu,Met239Lys,Val281Leu,E7:Glu292Ser,E8:Gly318stop)和第3外显子8对碱基的缺失,导致酶活性完全丧失则主要以失盐表现为主。
21-羟化酶、11β-羟化酶缺陷使醛固酮和皮质醇合成受阻,其前体堆积,向雄激素转化,过多的雄激素使女胚外生殖器男性化。若为酶完全缺陷,还可出现失盐症状。3β-类固醇脱氢酶缺陷使孕酮和17α-羟孕酮合成障碍,皮质醇、醛固酮及Δ4途径的雄激素合成受阻,但Δ5途径的17α-羟孕烯醇酮仍可向脱氢表雄酮转化,故其最终临床表现与21-羟化酶和11β-羟化酶缺陷相近,但患儿几乎恒定地出现失盐症状。17α-羟化酶缺陷使性激素及皮质醇合成受阻,男婴可出现女性外生殖器畸形,对女性性分化影响不大,但进入青春期后,因雌激素水平低下,女性表现为原发性闭经、子宫发育不良、第二性征发育差及FSH升高。
21-羟化酶、11β-羟化酶、3β-羟类固醇脱氢酶三者同时完全缺陷,患儿在出生后2周内即出现严重的水盐代谢紊乱症状,呕吐、腹泻、喂奶困难、哭声小、紫绀、脱水、萎靡、心律失常或心搏骤停而死亡,少有活过25岁者。三种酶的不完全缺陷,则出现女性假两性畸形,其外阴的表现可呈五种类型:①单纯阴蒂稍肥大;②阴蒂较肥大、阴道呈漏斗状、阴道口小,但开口和尿道口分开;③阴蒂显著肥大,前庭小,阴道和尿道共同开口;④阴蒂肥大,似阴茎、大阴唇部分融合如尿道下裂样表现;⑤完全男性化外阴、阴道与尿道共同开口,呈肥大的阴蒂头。三种酶不完全缺陷极轻症患者,性征可不受影响,仅因体内性激素紊乱,而出现原发闭经或一定程度的子宫发育不良,也可出现继发性闭经或月经稀发,青春期发病者临床上常难与PCOS区别。STAR位于人第8号染色体,编码34kD的线粒体磷酸化蛋白,促进胆固醇从线粒体膜外向膜内的转运,调节类固醇物质的应答急性反应,若该基因出现终止密码子前突变、框架移位突变、外来DNA序列插入,以及无功能的错义突变,则出现肾上腺皮质和卵巢甾体激素全面缺乏,患儿多于婴幼儿期夭折。若活到青春期后,女性则出现青春期延迟、原发闭经和Addison病的系列表现。男性则出现女性外生殖器、盲端阴道,无子宫,睾丸小且位于腹股沟或腹腔内。









