


 收藏
收藏 已收藏
已收藏英文名称 :Alzheimer's disease
阿尔茨海默病(Alzheimer's disease,AD)是发生于老年和老年前期、以进行性认知功能障碍和行为损害为特征的中枢神经系统退行性病变。临床上表现为记忆障碍、失语、失用、失认、视空间能力损害、抽象思维和计算力损害、人格和行为改变等。AD是老年期最常见的痴呆类型,约占老年期痴呆的50%~70%。随着对AD认识的不断深入,目前认为AD在痴呆阶段之前还存在一个极为重要的痴呆前阶段,此阶段可有AD病理生理改变,但没有或仅有轻微临床症状。
AD可分为家族性AD和散发性AD。家族性AD呈常染色体显性遗传,多于65岁前起病,最为常见的是位于21号染色体的淀粉样前体蛋白(amyloid precursor protein,APP)基因、位于14号染色体的早老素1(presenilin 1,PS1)基因及位于1号染色体的早老素2(presenilin 2,PS2)基因突变。携带有APP和PS1基因突变的人群几乎100%会发展为AD,而携带有PS2基因突变的人群,发展为AD的概率约为95%。对于占90%以上的散发性AD,尽管候选基因众多,目前认为载脂蛋白E(apolipoprotein E,APOE)基因最为有关。APOEε4携带者是散发性AD的高危人群,研究显示携带一个APOEε4等位基因的人群,其罹患AD的风险约为正常人的3.2倍,而携带有两个APOEε4等位基因的人群,其罹患AD的风险约为正常人的8~12倍。
有关AD的发病机制,现有多种学说,其中影响较广的有β-淀粉样蛋白(β-amyloid,Aβ)瀑布假说(the amyloid cascade hypothesis),认为Aβ的生成与清除失衡是导致神经元变性和痴呆发生的起始事件。家族性AD的三种基因突变均可导致Aβ的过度生成,是该学说的有力佐证。而Down综合征患者因体内多了一个APP基因,在早年就出现Aβ沉积斑块,也从侧面证明了该学说。另一重要的学说为tau蛋白学说,认为过度磷酸化的tau蛋白影响了神经元骨架微管蛋白的稳定性,从而导致神经原纤维缠结形成,进而破坏了神经元及突触的正常功能。近年来,也有学者提出了神经血管假说,提出脑血管功能的失常导致神经元细胞功能障碍,并且Aβ清除能力下降,导致认知功能损害。除此之外,尚有细胞周期调节蛋白障碍、氧化应激、炎性机制、线粒体功能障碍等多种假说。
AD发病的危险因素有低教育程度、膳食因素、吸烟、女性雌激素水平降低、高血压、高血糖、高胆固醇、高同型半胱氨酸、血管因素等。
AD是老年期最常见的慢性疾病之一,世界卫生组织(WHO)估计全球65岁以上老年人群AD的患病率为4%~7%,AD患病率与年龄密切相关,年龄平均每增加6.1岁,患病率升高1倍;在85岁以上的老年人群中,AD的患病率可高达20%~30%。2001年全球AD患者超过2000万,预计2040年将超过8000万。AD是造成老年人失去日常生活能力的最常见疾病,同时也是导致老年人死亡的第五位病因。AD不仅给患者带来巨大的痛苦,给家庭和社会也带来沉重精神压力和医疗、照料负担。2010年全世界用于AD的费用估计为6040亿美元。因此,AD已经成为影响全球的公共健康和社会可持续发展的重大问题。
AD的大体病理表现为脑的体积缩小和重量减轻,脑沟加深、变宽,脑回萎缩,颞叶特别是海马区萎缩(图1)。组织病理学上的典型改变为β淀粉样物质在神经细胞外沉积形成的神经炎性斑和过度磷酸化的tau蛋白在神经细胞内聚集形成的神经原纤维缠结,神经元缺失和胶质细胞增生(图2)。
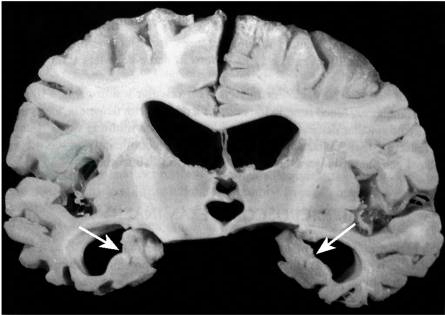
图1 阿尔茨海默病脑组织冠状切面
可见双侧海马明显萎缩,海马旁回变窄,侧脑室相应扩大
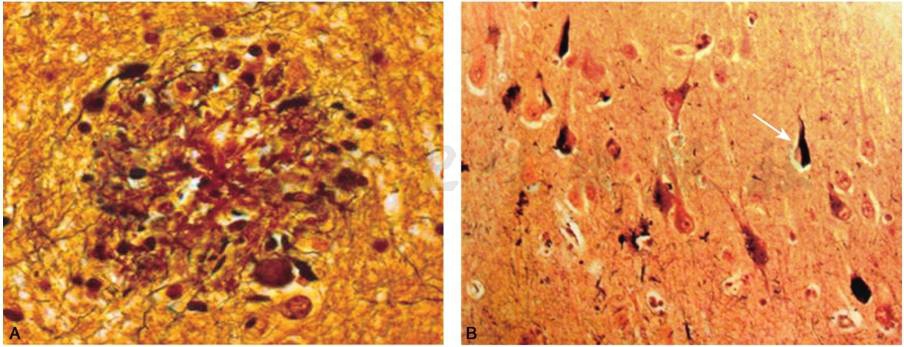
图2 阿尔茨海默病脑内病理表现
A.神经炎性斑;B.神经原纤维缠结(箭头)
1.神经炎性斑(neuritic plaques,NP)
在AD患者的大脑皮质、海马、某些皮质下神经核如杏仁核、前脑基底神经核和丘脑存在大量的NP。NP以Aβ沉积为核心,核心周边是更多的Aβ和各种细胞成分。自20世纪70年代以来,相继有研究者制定了诊断AD所需大脑皮质NP数量的神经病理诊断标准,目前广泛使用的是美国学者Mirra等1991年提出的半定量诊断标准,用图像匹配的方法估计三个脑叶新皮质严重受累区NP的数量。
2.神经原纤维缠结(neurofibrillary tangles,NFT)
大脑皮质和海马存在大量NFT,NFT主要在神经元胞体内产生,有些可扩展到近端树突干。含NFT的神经元细胞大多已呈退行性变化。NFT也常见于杏仁核、前脑基底神经核、某些下丘脑神经核、脑干的中缝核和脑桥的蓝斑。轻度AD患者,NFT可能仅限于内嗅皮质和海马。
AD的病理改变可能先于症状多年出现,即有病理改变存在而无认知受损的表现。病理改变和认知功能受损同时存在时,患者多为中度或重度的AD。如果认知受损的情况下仅仅观察到了轻度的AD病理改变,很可能存在其他疾病,不诊断AD。
1.实验室检查
血、尿常规,血生化检查均正常。CSF检查可发现Aβ42水平降低,总tau蛋白和磷酸化tau蛋白增高。
2.脑电图
AD的早期脑电图改变主要是波幅降低和α节律减慢。少数患者早期就有脑电图α波明显减少,甚至完全消失,随病情进展,可逐渐出现较广泛的θ活动,以额、顶叶明显。晚期则表现为弥漫性慢波。
3.影像学
CT检查见脑萎缩、脑室扩大;头颅MRI检查显示的双侧颞叶、海马萎缩(图3)。SPECT灌注成像和氟脱氧葡萄糖PET成像可见顶叶、颞叶和额叶,尤其是双侧颞叶的海马区血流和代谢降低。使用各种配体的PET成像技术(如PIB-PET、AV45-PET)可见脑内的Aβ沉积(图4)。
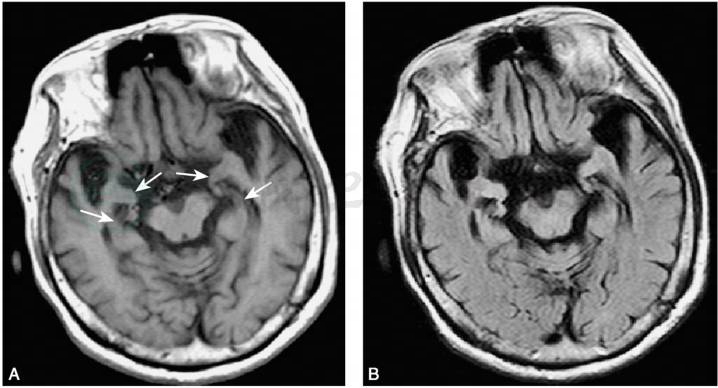
图3 MRI显示阿尔茨海默病颞叶和海马萎缩
A.T1加权像:双侧脑室颞角扩大,颞叶萎缩,以内颞叶、海马钩回萎缩明显(箭头);B.FLAIR像:萎缩的内颞叶、海马钩回呈高信号
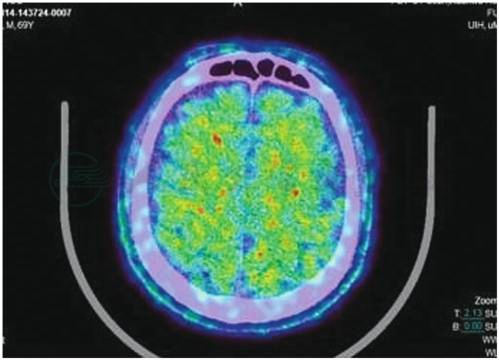
图4 18F-AV45 PET显示脑内Aβ沉积
4.神经心理学检查
对AD的认知评估领域应包括记忆功能、言语功能、定向力、应用能力、注意力、知觉(视、听、感知)和执行功能七个领域。临床上常用的工具可分为:①大体评定量表,如简易精神状况检查量表(MMSE)、蒙特利尔认知测验(MoCA)、阿尔茨海默病认知功能评价量表(ADAS-cog)、长谷川痴呆量表(HDS)、Mattis痴呆量表、认知能力筛查量表(CASI)等;②分级量表,如临床痴呆评定量表(CDR)和总体衰退量表(GDS);③精神行为评定量表,如汉密尔顿抑郁量表(HAMD)、神经精神问卷(NPI);④用于鉴别的量表,Hachinski缺血量表。还应指出的是,选用何种量表,如何评价测验结果,必须结合临床表现和其他辅助检查结果综合得出判断。
5.基因检查
有明确家族史的患者可进行APP、PS1、PS2和APOEε4基因检测,突变的发现有助于确诊和疾病的提前预防。
AD患者认知功能衰退目前治疗困难,综合治疗和护理有可能减轻病情和延缓发展。
1.生活护理
包括使用某些特定的器械等。有效的护理能延长患者的生命及改善患者的生活质量,并能防止摔伤、外出不归等意外的发生。
2.非药物治疗
包括职业训练、音乐治疗等。
3.药物治疗
(1)改善认知功能
①乙酰胆碱酯酶抑制剂(AChEI):包括多奈哌齐、卡巴拉汀、石杉碱甲等,主要提高脑内乙酰胆碱的水平,加强突触传递。②NMDA受体拮抗剂:美金刚能够拮抗N-甲基-D-门冬氨酸(NMDA)受体,具有调节谷氨酸活性的作用,现已用于中重度AD患者的治疗。③临床上有时还使用脑代谢赋活剂如奥拉西坦等。
(2)控制精神症状
很多患者在疾病的某一阶段出现精神症状,如幻觉、妄想、抑郁、焦虑、激越、睡眠紊乱等,可给予抗抑郁药物和抗精神病药物,前者常用选择性5-HT再摄取抑制剂,如氟西汀、帕罗西汀、西酞普兰、舍曲林等,后者常用不典型抗精神病药,如利培酮、奥氮平、喹硫平等。这些药物的使用原则是:①低剂量起始;②缓慢增量;③增量间隔时间稍长;④尽量使用最小有效剂量;⑤治疗个体化;⑥注意药物间的相互作用。
4.支持治疗
重度患者自身生活能力严重减退,常导致营养不良、肺部感染、泌尿系感染、压疮等并发症,应加强支持治疗和对症治疗。
目前,还没有确定的能有效逆转认知缺损的药物,针对AD发病机制不同靶点的药物开发尚处于试验阶段。处于AD痴呆前阶段的患者,宜饮食调整(地中海饮食)、体力锻炼和认知训练结合起来延缓认知功能下降。









