


 收藏
收藏 已收藏
已收藏英文名称 :polymyositis
多发性肌炎(polymyositis)是一组由多种病因引起的广泛的骨骼肌炎性病变,临床特点为急性、亚急性或慢性起病,对称性四肢近端和颈肌及咽肌无力,肌肉压痛,血清CK增高和病理提示骨骼肌纤维坏变及淋巴细胞浸润,同时可伴有血沉增快及肌电图呈肌源性损害,用糖皮质激素治疗效果好等特点。发病与细胞和体液免疫异常有关。本病的发病率为(0.1~0.9)/10万。
约半数多发性肌炎患者与HLA-DR3相关。HLA-DR52几乎见于所有的多发性肌炎患者,姐弟同患多发性肌炎的家族也不少见,说明遗传因素参与了发病。外因多与病毒感染和自身免疫功能异常有关。部分患者在发病前有流感病毒A和B、HIV、ECHO、柯萨奇病毒感染或寄生虫感染史,或有恶性肿瘤。有些患者合并红斑狼疮、类风湿性关节炎和硬皮病等。
多发性肌炎发病机制与免疫失调有关,包括细胞免疫和体液免疫的异常。90%的患者血清抗肌球蛋白抗体阳性;50%的患者抗核抗体阳性;肌纤维及其周围可见T辅助细胞;周围淋巴细胞对肌肉抗原敏感,并对肌细胞培养有明显的细胞毒作用,故本病是自身免疫性疾病。多发性肌炎以细胞免疫为主,与T细胞毒性淋巴细胞直接导致肌纤维的破坏,细胞间黏附分子、白细胞介素-1α和炎性细胞的浸润密切相关。目前尚不清楚什么因素直接诱发多发性肌炎的自身免疫异常,推测病原体感染改变了内皮细胞或肌纤维表面的抗原性,从而引发针对内皮细胞或肌细胞的免疫反应;或病毒感染后启动了机体对某些病毒肽段的免疫应答,而这些肽段与肌细胞中的某些蛋白的肽段结构相似,通过交叉免疫启动了自身免疫反应而攻击自身的肌细胞。体液免疫也参与了多发性肌炎的发病,抗体的作用机制可能为:①直接与肌膜上的靶抗原结合;②抗体与肌膜表面的蛋白呈交叉反应,引起组织损害;③补体参与引起免疫反应。
肌纤维呈角形、圆形或不规则形态;可见片状或散在肌纤维变性、坏死及吞噬现象;有较多的核内移肌纤维;肌内、外衣增宽。肌纤维间隙或肌束衣出现大量炎细胞浸润,炎性细胞围绕小血管分布。可有小动脉壁增厚,内皮细胞增生,甚至使管腔狭窄或完全闭塞。急性期还可出现肌纤维水肿空泡样变性及肌纤维溶解现象。慢性多发性肌炎伴有明显的肌纤维肥大、增生及分裂现象。严重者肌纤维数量明显减少,代以大量增生的结缔组织和脂肪组织。免疫组化染色提示的炎性细胞主要是单核细胞和CD8淋巴细胞;血管壁有免疫球蛋白和补体沉积;坏死肌纤维上有免疫补体C5~9沉积;未出现坏变的肌纤维的基质部和结缔组织中也有IgG沉积(图1)。
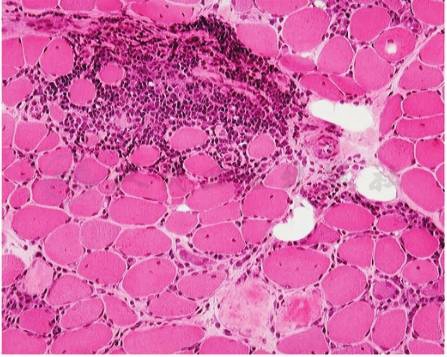
图1 多发性肌炎患者肌肉组织HE染色
肌纤维大小不等,萎缩肌纤维以小圆形为主,有些肌纤维出现坏变。上方有大量淋巴细胞浸润堆积于肌纤维间隙
1.急性期周围血WBC增高,血沉增快;血清CK明显升高,可达正常的10倍以上。1/3患者类风湿因子和抗核抗体阳性,免疫球蛋白及抗肌球蛋白抗体增高。24小时尿肌酸可增高。如合并横纹肌溶解者,可出现肌红蛋白尿。
2.肌电图可见自发性纤颤电位和正锐波,多相波增多,运动单位电位时限缩短和波幅降低等肌源性损害的表现。神经传导速度正常。
3.52%~75%的患者有心电图异常,QT延长、ST段下降。
4.肺部X线或CT检查可发现肺部片状阴影。
5.肌活检是诊断与排除其他肌病的重要手段。病理改变见前文所述。
急性期患者应卧床休息,适当活动以保持肌肉功能和避免挛缩,注意防止肺炎等并发症。
1.肾上腺皮质激素 为首选药物,且应该进行首次或早期冲击治疗,效果更佳。甲泼尼龙琥珀酸钠1000mg,静脉滴注,每日1次,连用3~5天,随后每日逐减半量,即500mg、250mg、125mg,继之改为口服醋酸泼尼松(泼尼松)60mg,最后酌情逐渐减量;地塞米松20mg,静脉滴注,每日1次,连用1周,随后改为口服醋酸泼尼松并酌情逐渐减量至维持量;有的患者可直接给予口服醋酸泼尼松60~100mg,每日早顿服,连续10天后,开始酌情减量至维持量。多数患者在激素冲击治疗后一周左右症状开始减轻,6周左右症状明显改善,然后持续8~12周后逐渐减量,醋酸泼尼松的维持量因人而异,一般为5~20mg,可应用1~3年。如果在减量过程中或应用维持量过程中出现病情复发加重,则重新采用大剂量冲击。长期皮质类固醇激素治疗应注意预防副作用,给予低糖、低盐和高蛋白饮食,用抗酸剂保护胃黏膜,注意补充钾和维生素D,对结核病患者应进行相应的治疗。
2.静脉注射免疫球蛋白 有条件者可为首选治疗办法,且有较好的效果。丙种免疫球蛋白,0.4g/(kg·d),静脉滴注,每次连续3~5天,每月可重复一次,连续3~5个月。
3.免疫抑制剂 在激素治疗不满意时加用。可选用其中一种,如甲氨蝶呤、硫唑嘌呤、环磷酰胺、环孢素,用药期间注意定期查白细胞和肝肾功能。
4.血浆置换 泼尼松和免疫抑制剂治疗无效并伴有明显吞咽困难、构音障碍者可用血浆置换治疗,以去除血液中的淋巴因子和循环抗体,可改善肌无力的症状。
5.给予高蛋白和高维生素饮食,进行适当体育锻炼和理疗。重症者应预防关节挛缩及失用性肌萎缩。









