


 收藏
收藏 已收藏
已收藏英文名称 :muscular dystrophy
肌营养不良症(muscular dystrophy)是一大类与遗传有关的肌纤维变性和坏死性肌病。表现为进行性肌肉无力和萎缩。肌营养不良症现根据肌肉无力的分布、起病年龄、遗传方式、致病的基因突变和异常基因产物来分类(表1)。
表1 部分肌病的致病基因突变和异常基因产物
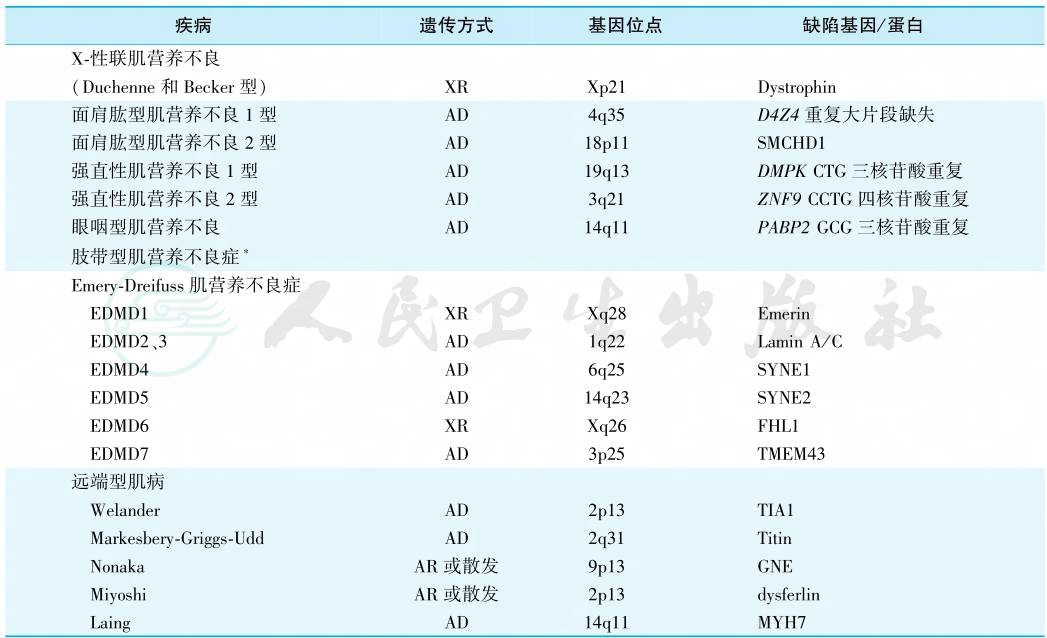
注:AD.常染色体显性遗传;AR.常染色体隐性遗传;GNE.UDP-N-乙酰氨基葡萄糖2-表位酶/N-乙酰甘露糖胺激酶;XR.X连锁隐性遗传。
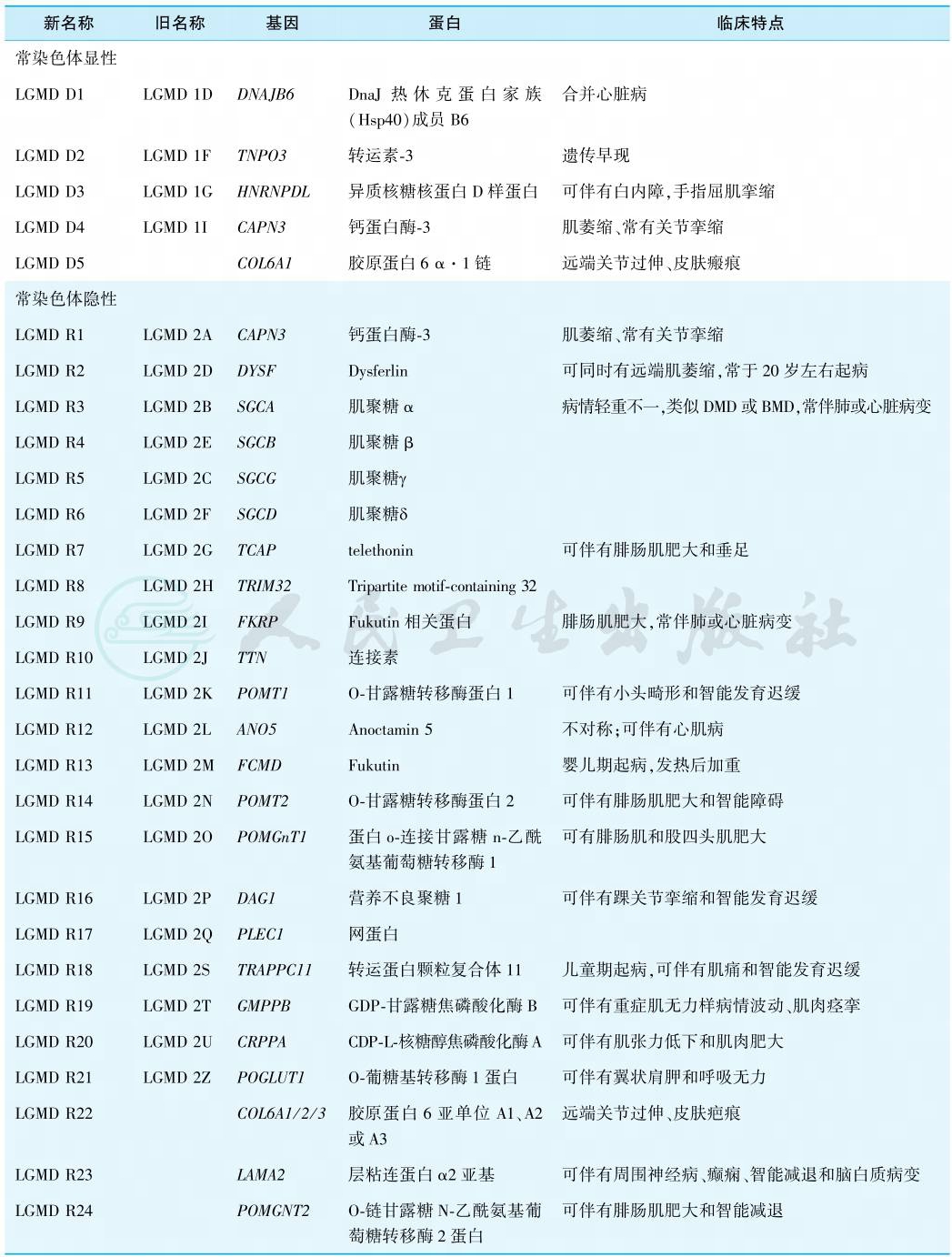
肢带型肌营养不良(limb girdle muscular dystrophy,LGMD)包括一大类肩带和骨盆带肌无力呈肢带型分布模式的遗传性肌营养不良,其中包括常染色体隐性遗传(占绝大多数)和常染色体显性遗传两大类。肢带型肌营养不良在男女中的发病概率相等,如果发生在儿童早期,其临床表现与Duchenne型肌营养不良相似;较轻的表型可类似于Becker型肌营养不良。实验室检查结果(血清肌酸激酶、肌电图、肌肉活检)符合肌营养不良的特点(表2)。目前已有数十种肢带型肌营养不良亚型根据基因突变和其导致的蛋白缺陷而命名,由于新的亚型不断被发现,目前对其命名规则进行了改变(表2),较为少见的常染色体显性LGMD命名为D型(LGMDD1、D2、D3等),常染色体隐性 LGMD 命名为 R 型(LGMDR1、R2等)。
表2 LGMD的新旧命名和临床特点
抗肌萎缩蛋白病(dystrophinopathy)为肌营养不良症中最常见类型,亦称为假肥大型肌营养不良症,主要包括Duchenne型、Becker型和女性携带者。参照国外资料,Duchenne型肌营养不良的发病率在男婴中约为1/3500。
一、病因与发病机制
假肥大型的Duchenne型肌营养不良和Becker型肌营养不良均为Xp21上dystrophin基因缺陷所致,属X连锁隐性遗传。破坏了基因翻译阅读框的突变(移码突变)导致抗肌萎缩蛋白完全缺失,造成Duchenne型肌营养不良。而不改变翻译阅读框的框内突变则会翻译表达出数量或大小异常的抗肌萎缩蛋白,仍能保持其部分功能,导致Becker型肌营养不良。
二、病理
呈现肌营养不良的特征性病理改变,免疫组化提示肌膜的抗肌萎缩蛋白完全或部分缺失。
三、临床表现
呈X-性连锁隐性遗传,男性患病,女性携带。
1.Duchenne型病情较重 通常在幼儿期起病。表现为学步困难、易跌、跌倒后不易爬起。臀中肌受累而致骨盆左右上下摇动,跟腱挛缩而足跟不能着地,腰大肌受累而腹部前凸;头后仰,呈“鸭步”。从平卧位起来有“Gowers现象”。
继骨盆带肌受累之后,逐步出现肩胛带肌萎缩、无力,双臂上举不能,肩胛骨可呈翼状耸起,称“翼状肩”。多数患者有腓肠肌肥大,病初肥大肌肌力可相对较强。
病程逐步发展。少数儿童由于本身生长发育的影响,可能出现病程相对稳定或好转。多数病孩到10岁已丧失行走能力,依靠轮椅或坐卧不起,出现脊柱和肢体畸形。晚期四肢挛缩,活动完全不能。常因伴发肺部感染、褥疮等。于20岁之前夭折。
约20%的患者有不同程度智能减退。多数患者可有心肌损害,早期可无症状,晚期可出现心功能衰竭。
2.Becker型 常于12岁左右起病,受累肌群的分布、假肥大和心电图异常与Duchenne型相似,但程度相对较轻。部分患者即使在晚年也没有明显症状,预期寿命略低于正常人。
3.女性携带者所生育的男性后代有50%可能患病,女性后代有50%可能成为携带者。女性携带者通常无症状,但少数情况下也会出现轻度肢带肌无力,50%的女性携带者肌酸激酶水平会升高。
用羊水脱落细胞或绒毛膜可以进行产前遗传检测。
四、诊断与鉴别诊断
依据男性患儿、进展性肢带肌无力、腓肠肌肥大和高肌酸激酶血症等临床特征可高度拟诊。确诊需依赖于病理免疫组化和基因分析。临床上需与肢带型肌营养不良和脊肌萎缩症相鉴别。
五、治疗
口服泼尼松推荐的起始剂量为0.75mg/(kg•d),疗效包括延长行走能力保留的时间2年。基因治疗已经取得了初步进展,Eteplirsen是首个获批治疗DMD的药物。采用了寡核苷酸和外显子跳跃技术,目的是修复mRNA的阅读框来部分纠正遗传缺陷,通过跳跃51号外显子,产生一种较短但仍具有功能的抗肌萎缩蛋白,从而稳定或减缓疾病的进程,延长并改善DMD患者的生活质量。此外,Ataluren/PTC124获得欧盟药监当局的有条件上市批准,用于5岁及以上无义突变型杜氏肌营养不良非卧床患者的治疗。Ataluren是一种蛋白质修复药物,用于由无义突变所致遗传性疾病的患者群体,选择性促进核糖体提前终止密码子而不是正常终止密码子的通读,旨在使含有无义突变的基因,产生功能蛋白。
面肩肱型肌营养不良是一种常染色体显性遗传病,外显率高,在同一家族内表现轻重不一。发病率约为1/20000。
一、病因与发病机制
面肩肱型肌营养不良呈常染色体显性遗传,大部分患者是由于染色体4q35亚端粒区D4Z4大片段重复缩短致病,导致该染色体区域变为低甲基化的疏松状态,进而表达促凋亡因子DUX4蛋白。此类患者约占所有FSHD患者的95%,称为面肩肱型肌营养不良1型(FSHD1)。此外,染色体结构维持蛋白SMCHD1突变可导致染色体甲基化程度降低,这样所导致的FSHD称为面肩肱型肌营养不良2型(FSHD2)。
二、病理
肌肉病变较轻,肌纤维大小不一,仅少数纤维坏死与再生。可见很小角状纤维,无群组化,ATP酶染色纤维分型正常。肌纤维间和肌束间可见灶性炎症细胞浸润,为炎症细胞对坏死肌纤维的反应。
三、临床表现
两性罹病概率相等,自婴儿至中年均可起病。婴儿期的闭眼不全可能不被家长注意。主要临床表现为眼睑闭合无力,皱额、鼓腮、吹哨和露齿不能或无力,重者呈面具状脸。嘴唇肥厚而微翘。颈部胸锁乳突肌明显萎缩或变细,两臂平举起时可见颈肌悬吊肩胛而呈特殊的“蝠翼状”。肩胛带肌肉明显萎缩;胸大肌萎缩内陷,锁骨水平支撑,肩胛部呈现“衣架肩”。两下肢受累较轻。部分患者腹部Beevor征阳性。本病发展缓慢,常有顿挫或停止发展。
四、诊断与鉴别诊断
依据典型常染色体显性遗传,面肌、肩、肱肌和踝背屈肌无力伴萎缩,临床诊断一般不难。此外,肌肉无力萎缩不对称、肌群由上而下逐步受累、三角肌相对保留、伴有高频听力丧失或视网膜血管病变等也是诊断本病的重要依据。但临床上仍需与肢带型肌营养不良症、肩腓综合征等鉴别。
五、治疗
主要是支持治疗。对于翼状肩胛明显并手举过头困难的患者,可通过外科手术固定肩胛骨以改善症状。沙丁胺醇和一水肌酸可能会改善患者的肌力,但有待进一步证实。
强直性肌营养不良症(myotonic muscular dystrophy,DM)是一组以肌强直、进行性肌萎缩、白内障、心脏传导阻滞、性腺萎缩以及智能低下为主要特点的多系统疾病。目前分为强直性肌营养不良1型和2型(DM1型和DM2型)。DM1是最为常见的肌营养不良之一,发病率为13.5/10万新生儿,患病率为(3~5)/10万。
一、病因与发病机制
强直性肌营养不良1型与19q13上肌强直蛋白激酶(DMPK)基因3’端非翻译区的CTG三核苷酸过度重复有关,疾病严重程度随重复拷贝数不同而有差异。正常个体拷贝数为5~30个。CTG拷贝数从双亲到子代有扩增趋势,因此本病有遗传早发现象。DM2与 3q21号染色体上锌指蛋白9(ZNF9)基因的CCTG四核苷酸过度重复有关。
二、临床表现
DM1的临床表型跨度较大,可从无症状到重症致死先天性,而DM2则相对症状较轻。DM1根据发病年龄可分为5型(表3),其中经典型最为常见,可累及所有系统。
表3 强直性肌营养不良1型的临床分型
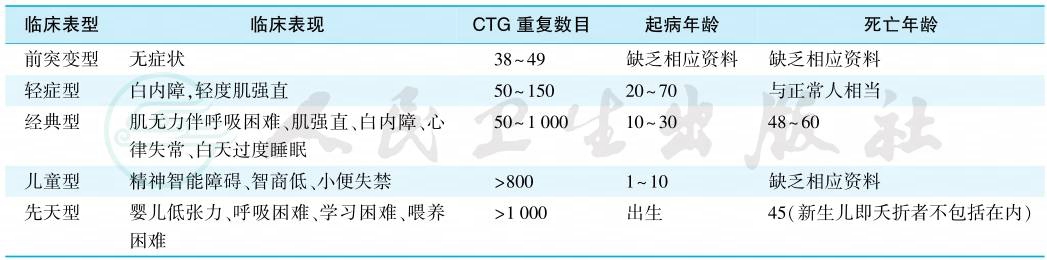
经典型DM1常在10~30岁起病,主要特征为受累骨骼肌肉萎缩、无力和肌强直,以前两种症状为突出。强直症状在重复运动后可有缓解。随着肌萎缩的进展,肌强直可消失。体格检查可见唇微翘,颧骨隆起、额肌萎缩而呈斧头状(图1);颈细长,胸锁乳突肌萎缩而头部前倾伸长,称鹅颈;构音不清或伴吞咽困难,握拳后不能立即放开,手指不能伸直。叩诊槌叩击被检肌肉时出现肌球。

图1 强直性肌营养不良1型患者唇微翘、颧骨隆起、额肌萎缩
此外,由于累及心脏、眼部、消化道、内分泌及神经系统等,多数患者出现不同程度心脏传导阻滞、白内障、腹痛、腹泻、性欲减退、早秃、糖尿病、认知障碍和人格改变等,头颅MR可见脑白质多发病变,尤其以颞叶前部最易累及,同时伴有脑萎缩和脑室扩大。39%患者出现日间过度睡眠。女性患者流产率高。
DM2多于30岁后起病,多系统受累与DM1极为相似,其中以心脏传导阻滞、白内障和胰岛素抵抗较为常见,认知功能受损轻于DM1。DM2骨骼肌受累相对较轻,主要累及近端肌肉和颈部屈肌,肌痛和僵硬较DM1常见,但预后相对较好。
三、诊断与鉴别诊断
依据典型的肌强直、进行性肌萎缩和多系统损害的临床特征,一般不难诊断。但亚型的确诊需依赖于基因分析。
四、治疗
本病尚无特效治疗,目前使用反义寡核苷酸在体外和动物治疗中取得了良好的疗效,有待进一步临床验证。临床上主要以支持对症治疗为主,针对受累的不同系统分别给予对症支持治疗,譬如踝脚矫形器改善足下垂,手夹板改善手功能;植入心脏起搏器治疗心脏传导阻滞;无创呼吸机或机械通气改善呼吸困难;口服降糖药或胰岛素控制血糖;眼科手术植入人工晶体治疗白内障;苯妥英钠、美西律和乙酰唑胺改善肌强直症状;莫达非尼改善DM1的日间多睡等。对于晚期严重患者,呼吸训练和体位引流法可以避免肺部感染。
Emery-Dreifuss肌营养不良症(Emery-Dreifuss muscular dystrophy,EDMD)既往认为系X连锁遗传,随后发现还有常染色体显、隐性遗传者,目前认为有6个基因与之有关(表1)。该病的特点是三大临床主征:①早期出现肘、踝关节的挛缩;②四肢无力缓慢进展,通常呈肩肱腓型分布;③心肌病伴房性传导阻滞。早期出现肘关节挛缩经常是导致诊断的关键特征(图2)。本病的临床诊断要点是上述三联征,有家族史则更为支持。但仍需与某些先天性肌病、肢带型肌营养不良症相鉴别。确诊需依赖于活检病理的特殊免疫染色和基因诊断。本病进展缓慢,很少有丧失行走能力者。目前尚无特效治疗,矫形治疗是解决关节挛缩并恢复关节功能的重要手段。患者多伴有心脏问题,及时评估心脏情况和放置起搏器是改善预后之关键。
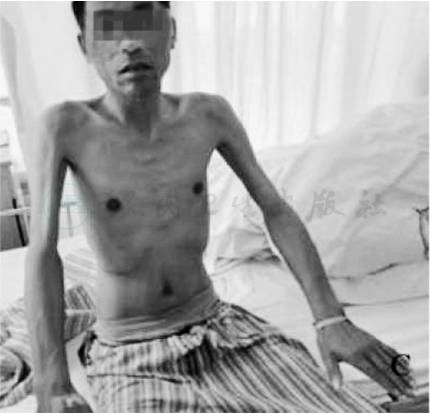
图2 Emery-Dreifuss肌营养不良患者示肩、上臂等肌群挛缩









