


 收藏
收藏 已收藏
已收藏英文名称 :hereditary ataxia
小脑的损害的主要表现是小脑性共济失调,核心症状是平衡障碍,步态异常,双手动作笨拙和构音障碍。
遗传性共济失调(hereditary ataxia)是指由遗传所致的以小脑性共济运动障碍、辨距不良为主要临床表现的一大类中枢神经系统变性疾病。临床除小脑损害表现外,可合并多系统损害。此外,遗传性共济失调尚可存在血液系统疾病(低β脂蛋白血症、棘红细胞增多症等)、代谢异常(维生素E、辅酶Q10异常及甲羟戊酸尿等)和生殖系统损害。遗传性小脑共济失调根据遗传特征分为常染色体显性遗传性共济失调、常染色体隐性遗传性共济失调、X连锁遗传性共济失调及带有线粒体异常的遗传性共济失调。本组疾病占神经遗传病的10%~15%。
遗传性共济失调按照发病年龄分类:一类是20岁以前发病,多为常染色体隐性遗传,称为早发性遗传性共济失调;另一类是20岁以后发病,多为常染色体显性遗传,称为晚发性遗传性共济失调。
一、早发性遗传性共济失调
Friedreich共济失调(Friedreich ataxia,FRDA)由 Friedreich首先于1863年报道,在西方国家的遗传性共济失调中约占半数。
二、晚发性遗传性共济失调
晚发性遗传性共济失调(late-onset inherited ataxias)是遗传性共济失调的主要类型,多数呈常染色体显性遗传,患病率约为(8~12)/10万。目前按照疾病基因被确定的先后次序命名,即数字标识的脊髓小脑性共济失调(spinocerebellar ataxia,SCA),如SCA3。现已确定的SCA基因类型迅速增加,已确认超过40个亚型(其中有部分为等位基因变异,如SCA15/SCA16)。
一、早发性遗传性共济失调
FRDA为常染色体隐性遗传,致病基因为位于9q21.11的FXN。除少数(1%~3%)为点突变或缺失外,95%的FRDA为该基因1号内含子上的一段GAA重复序列发生异常扩增,病理性抑制其编码蛋白frataxin蛋白,损害线粒体呼吸链中亚基铁硫簇生物发生,破坏线粒体铁稳态,造成心脏、神经系统和胰腺β细胞的毒性。
二、晚发性遗传性共济失调
SCA的突变类型中最常见的是重复序列拷贝数异常增加,包括编码区核苷酸 CAG重复扩增(SCA1、SCA2、SCA3、SCA6、SCA7、SCA17、DRPLA),非编码区 CAG 重复扩增(SCA12),其他重复扩增(SCA8、SCA10、SCA31、SCA36、SCA37)和其他类型突变。 SCA1、SCA2、SCA3、SCA6、SCA7、SCA17 及 DRPLA 这 7种亚型的致病基因的编码区域内都有一段CAG重复序列,当发生异常扩增,拷贝数超过一定范围时可导致基因的编码蛋白产生多聚谷氨酰胺扩展突变。此种突变造成的疾病统称为多聚谷氨酰胺疾病(poly glutamine disease,Poly Q disease)。Poly Q疾病有一个显著的共同特点为遗传早现(anticipation)现象,即在遗传性共济失调家系的连续几代人中,发病年龄逐代提前,症状逐代加重。
遗传性共济失调最早分为Friedreich和Marie型共济失调。1861年Friedreich首先报告一组少年发病,以常染色体隐性遗传为主,病变主要在脊髓,小脑亦有改变的疾病,后称为Friedreich共济失调(Friedreich ataxia,FRDA),该病名一直沿用至今。1893年Marie报告的一组病例比Friedreich共济失调发病晚,呈常染色体显性遗传,以小脑共济失调为主,亦有脑干、脊髓症状,并有视力障碍,被称为Marie型共济失调。Déjèrine和Thomas于1900年引入了OPCA的概念。50年后Greenfield依据病理解剖的特点将遗传性共济失调分为三类:以脊髓型为主的遗传性共济失调、以脊髓小脑型为主的遗传性共济失调及以小脑型为主的遗传性共济失调。对于临床医生来说,对患者进行诊断才是关键的,因此这种病理分型并不实用。几经更改,Harding在1993年按照临床表现和病理特征对常染色体显性遗传性共济失调(autosomal dominat cerebeller ataxia,ADCA)进行了分类:ADCAⅠ,小脑性共济失调,伴其他神经系统症状,包括视力萎缩、眼肌麻痹、锥体系和锥体外系征、外周神经病、痴呆等。SCA1-4、SCA17、SCA21、DRPLA等属于此型。ADCAⅡ,小脑性共济失调伴视网膜病变为特征,SCA7属于此型。ADCAⅢ,表现为纯小脑性共济失调,肌张力反射增强、凝视眼球震颤、面部肌肉持续颤动、震动感觉下降等,很少出现眼肌麻痹、四肢痉挛等,SCA5、SCA6、SCA8、SCA10、SCA12等属于此型。
随着分子生物学技术的飞速发展,SCA被定义成为一种单基因遗传病。1993年Orr等人将SCA1的致病基因定位在6p22-23并克隆了致病基因。至此,SCA的概念真正登上了历史舞台。从1993年至今,不断有新的SCA亚型的致病基因被定位及克隆。因此,一种以遗传学为基础的基因型分型应运而生。根据研究者对于致病基因定位的时间顺序,由国际人类基因组组织命名委员会[The Human Genome Organisation(HUGO)Gene Nomenclature Committee]进行命名。目前已经定位或克隆了将近50种SCA亚型。随着研究的深入,会不断有新的SCA亚型被发现,因而此种分型方法相对其他方法来说可谓顺应研究发展的潮流,充满生命力。
对遗传性共济失调的分型目前国际上较为通用的是按照遗传方式分为四种:常染色体显性遗传性共济失调、常染色体隐性遗传性共济失调(autosomal recessive cerebeller ataxia)、X连锁遗传性共济失调(X-linked hereditary ataxias)及带有线粒体异常的遗传性共济失调(ataxias with mitochondrial disorders),临床上以前二者多见(表1)。其中常染色体显性遗传性共济失调又可分为:SCA各亚型、DRPLA及EA(表2),常染色体隐性遗传性共济失调可分为FRDA、共济失调毛细血管扩张(ataxia telangiectasia,AT)等(表3)。
表1 遗传性共济失调的分型

表2 常染色体显性遗传性共济失调的基因型分型及各型临床特点

续表
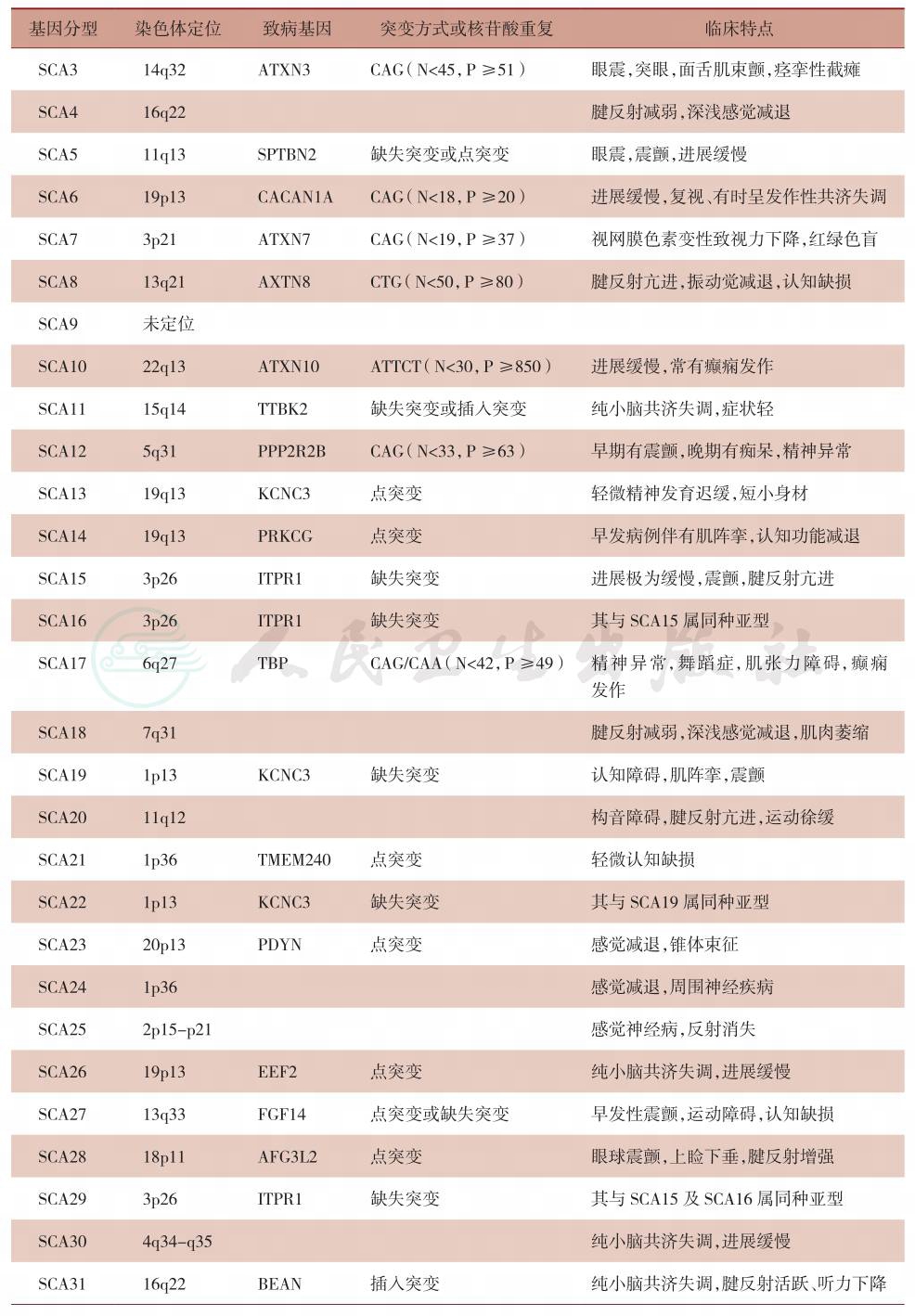
续表
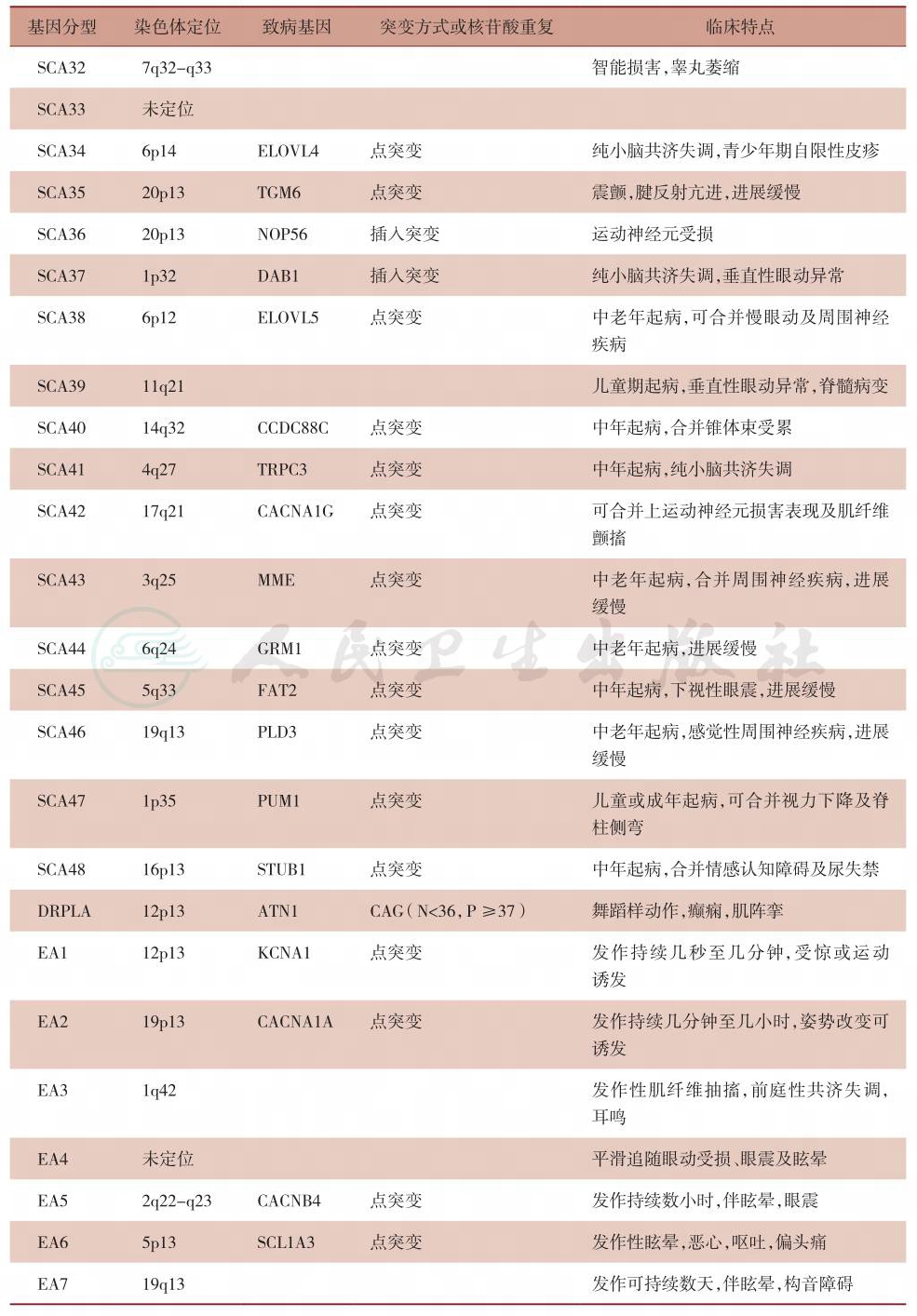
表3 常见常染色体隐性遗传性共济失调的基因型分型及各型临床特点
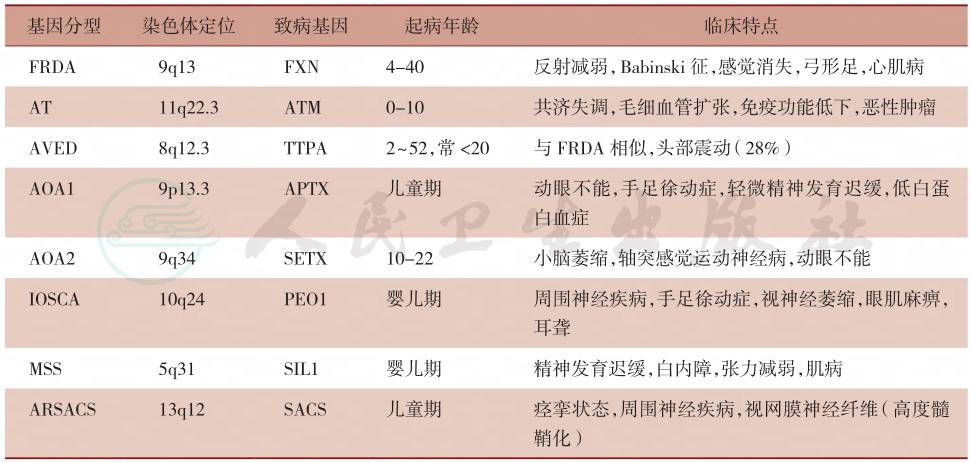
一、早发性遗传性共济失调
小脑齿状核和小脑上脚萎缩明显。脊髓变细,胸段最突出,脊髓后索、脊髓小脑束、锥体束均可见髓鞘脱失和轴突变性。心肌病是本病的特征之一,呈进行性的心肌肥厚、慢性间质性纤维变性和炎性浸润的表现。
二、晚发性遗传性共济失调
肉眼可见小脑半球和蚓部萎缩,脑干萎缩变小,以脑桥及下橄榄核明显;脊髓的颈段和上胸段明显萎缩。镜下主要为小脑、脑桥、下橄榄核萎缩,细胞脱失伴胶质增生。
一、早发性遗传性共济失调
除本病目前尚无特效治疗,FRDA的复杂和可变的临床表型需要广泛的多学科管理方法。物理治疗和对严重脊柱侧凸者手术矫正可改善生活质量。
二、晚发性遗传性共济失调
目前尚无特效治疗。康复治疗和经颅磁刺激是少数认为对小脑共济失调有帮助的治疗方法。









