


 收藏
收藏 已收藏
已收藏英文名称 :anhidrotic ectodermal dysplasia with immunodeficiency
NF-κB抑制子激酶γ(IKKγ,IKBKG),又称为NF-κB重要调节子(NEMO),其突变引起X连锁隐性的无汗性外胚层发育不良伴免疫缺陷病1型(anhidrotic ectodermal dysplasia with immunodeficiency type 1,EDID1),EDID2型由常染色体显性NF-κB抑制子α(inhibitor of NF-κB α,IKBA)突变引起。患儿具有可变的外胚层发育不良特征,但大部分包括少汗/无汗,严重度可变的免疫和感染表型。
NF-κB是根植于很多受体通路的核效应子,包括炎症性肿瘤坏死因子和Toll样受体超家族,功能为调节基因转录。NF-κB存在于细胞质,与NF-κB抑制子(IκB)结合。IκB激酶(IKK)通过磷酸化IκB和泛素化使NF-κB得以释放。IκB最终被蛋白酶体水解。目前认为由两种不同的信号通路活化NF-κB:经典和非经典通路。在很多细胞,促炎刺激如脂多糖、TNF和IL-1是NF-κB活性的强力诱导剂。大部分受体与配体结合后,通过经典途径活化NF-κB。受体积聚后利用大量的信号体诱导IKK复合体的积聚。IKK复合体至少包括IKKα、IKKβ、IKKγ。IKKγ是调节单元,不具有激酶活性。IκB蛋白被经典IKK复合体在2个特异的N端丝氨酸磷酸化,然后作为含有转导重复蛋白β泛素连接酶(β-transducingrepeat-containing protein,β-TrCP)的锚平台。泛素化(赖氨酸-48连接的链)诱导蛋白酶体介导的IκB蛋白降解,不影响结合的NF-κB二聚体的完整性。释放的NF-κB二聚体可结合DNA和活化基因。仅一部分肿瘤坏死因子受体超家族成员如CD40、BAFF-R、淋巴毒素受体β(LTβR)与配体结合后利用非经典通路。这些受体使信号体积聚,利用诱导NF-κB的激酶,导致IKKα依赖的IKKγ非依赖的过程,产生来源于P100的P52与RelB的二聚体。经典通路参与调节免疫反应过程中炎症和淋巴细胞的增殖和凋亡,非经典通路与淋巴器官的发育有关,确保产生足够的抗体反应。
通过对色素失禁症(incontinentia pigmenti,IP)的研究发现引起外胚层发育不良的基因与IKK的特异联系。IP是在妇女中出现的皮肤病,表现为色素沉着和其他外胚层缺陷。男性出生前是致死性的,结合明显受累的女性染色体偏移灭活明显,导致推测受累基因位于X染色体。IP中的NEMO突变典型是大的缺失,也包括移码突变和无义突变,引起蛋白明显改变导致疾病。由于偏移的X染色体灭活使免疫细胞中正常的NEMO存在允许正常的免疫能力。由于IP中的外胚层表现与外胚层发育不良(ectodermal dysplasia,ED)中的相似,外胚层发育不良蛋白(ectodysplasin,EDA)是TNF超家族成员,因此NEMO的特异评估是相关性的。NEMO突变的不典型IP妇女生育的男性具有EDID。EDID患儿具有类似于X连锁高IgM综合征的体液免疫缺陷,TNFR超家族成员CD40配置不反应。这些相关性导致研究集中于TNFR下游信号,最终NEMO基因突变被发现。
由于CD40信号被B细胞共刺激所需和依赖于NEMO活化的NF-κB,NEMO缺陷患儿感染的高敏感性部分是由于特异抗体产生缺陷伴或不伴有低丙种球蛋白血症。T细胞受损可能与分枝杆菌病有关。BCL10在TLR信号中被NF-κB活化所需。BCL10与NEMO(K399)区域结合,该区域经常被致病突变所改变。NEMO突变患儿NK细胞功能缺陷,在NK细胞毒过程中需要NF-κB活化。核苷酸结合寡聚结构域2(NOD2)信号依赖NF-κB,克罗恩病(Crohn’s disease)相关的NOD2突变不能传递正常信号,提示NEMO缺陷患儿炎性肠病的发病机制可能与此相关。
BCR和TCR利用非直接的由CARD11/BCL10/MALT-1复合体组成的控制的信号体通路通过IκBα进行信号传导。所有IκBα缺陷患儿细胞显示针对各种不同表面受体刺激的NF-κB反应受损,包括TLR、IL-2R、TCR和BCR,参与细胞内在的、天然的和获得性免疫。一旦IκBα的S32或S36丝氨酸残基被磷酸化,降解体变为β-TrCP的底物,后者是Skp-1-Cullin-1-F-box蛋白酶体降解复合体的成分,后者添加多泛素链(K48)至赖氨酸K21和K22,使靶IκBα降解。所有突变通过阻断Ser32或Ser36位残基的磷酸化和后续的降解来增强IκBα的抑制活性。
IKBKG(NEMO)位于Xq28,具有10个外显子,编码419位氨基酸的蛋白质。NEMO蛋白具有一系列重要的结构域:N端卷曲-卷曲1(curling-curling 1,CC1)结构域,中间的螺旋-环-螺旋2(helix-loop-helix 2,HLX2)结构域,C端CC2-亮氨酸拉链(leucine zipper,LZ)结构域,C端锌指(zinc finger,ZF)结构域。也作为假基因存在,但仅具有外显子3-10,不产生转录和不具有功能意义。
NEMO的功能依赖于二聚体化和其与线性的或K63连接的多泛素化链相互作用能力有关。这个能力需要CC2-LZ结构域和ZF结构域,前者参与二聚体化及包含一个泛素结合位点被称为NOA/UBAN/NUB,后者包含第二个泛素结合位点。NEMO基因P. Ala288Gly突变使NEMO二聚体不稳定,影响IκB激酶复合体的组装。P. Asp311Asn,p. Asp311Gly突变损伤NEMO-泛素结合。P. Glu315Ala,p.Arg319Gln突变破坏Glu315和Arg319间的盐桥的形成,p. Glu315Ala的折叠缺陷使NEMO不能与泛素链连接。P. Cys417Phe突变修饰C端ZFα螺旋的结构,降低其稳定性。P. Cys417Arg突变损伤针对CD40L的c-Rel活化。
53%NEMO基因突变影响外显子10,占所有患儿的62%。最常见的是位于1 161~1 167间的胞嘧啶束的插入突变,或累及417位丝氨酸的错义突变。大部分突变影响C端锌指结构域。所有已知的外显子10的突变导致外胚层发育不良。产生延长蛋白的突变(c.1259A>G)也导致骨硬化和淋巴水肿。影响外显子4~9的突变也被描述,位于外显子8的突变最常见。4例患儿具有免疫缺陷(immunodeficiency,ID),但不具有ED,均累及外显子9或外显子8的3′区域,这些区域编码亮氨酸拉链或其周围区域,也包括改变E315和P373间的NEMO蛋白的突变。深的内含子突变IVS4+866C>T被报道。在NEMO的潜在锌指结构域的Cys417Arg和 Asp406Val突变导致X-连锁高IgM伴外胚层发育不良(X-linked hyper IgM with ectodermal dysplasia,XHMED)。NEMO 基因 c.518C > G(Arg173Gly)突变引起孤立反复的侵袭性肺炎链球菌感染。
女性患儿染色体灭活从随机变为偏移,伴随免疫缺陷消失。一个NEMO突变女性具有EDID,与随机的非偏移的X染色体灭活有关(图1)。孤立的隐匿的牙齿异常见于3例具有严重免疫缺陷的携带者,2例X染色体随机灭活,1例偏移灭活。一个男性具有严重的NEMO突变,原因为合子后的体细胞嵌合允许存活。其他具有严重NEMO突变的男性,或者通过体细胞嵌合或者具有Klinefelter综合征(47XXY),具有IP,但不具有EDID。
IκBα是丝氨酸/苏氨酸蛋白激酶家族一员,包含N端的磷酸化位点,中间锚蛋白重复结构域,在C端是富含脯氨酸、谷氨酸、丝氨酸和苏氨酸的重复的肽序列。11种不同的突变被鉴定,均影响外显子1编码的前76位N端氨基酸,包括错义突变(S32I、S32G、S32R、S32N、G33V、S36Y、M37K、M37R)和无义突变(Q9X、W11X、E14X)。P. Ser32Ile突变取消了IKBA基因Ser32位点的磷酸化。P. Gln9X、p. Glu14X、p. Trp11X突变重新启动起始位点Met37,产生缺乏关键的丝氨酸磷酸化残基32和36的N端截断蛋白。P. Ser36Tyr突变导致IκBα降解缺陷。P. Met37Lys突变由于IκBα蛋白获得性功能阻断NF-κB的活化。
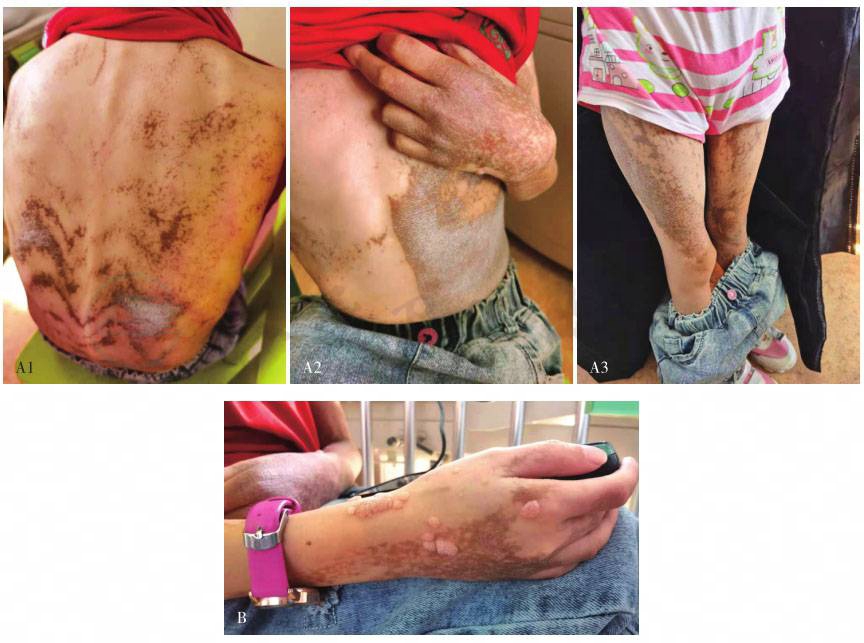
A 皮肤色素失禁症表现;B 手部皮肤多发疣
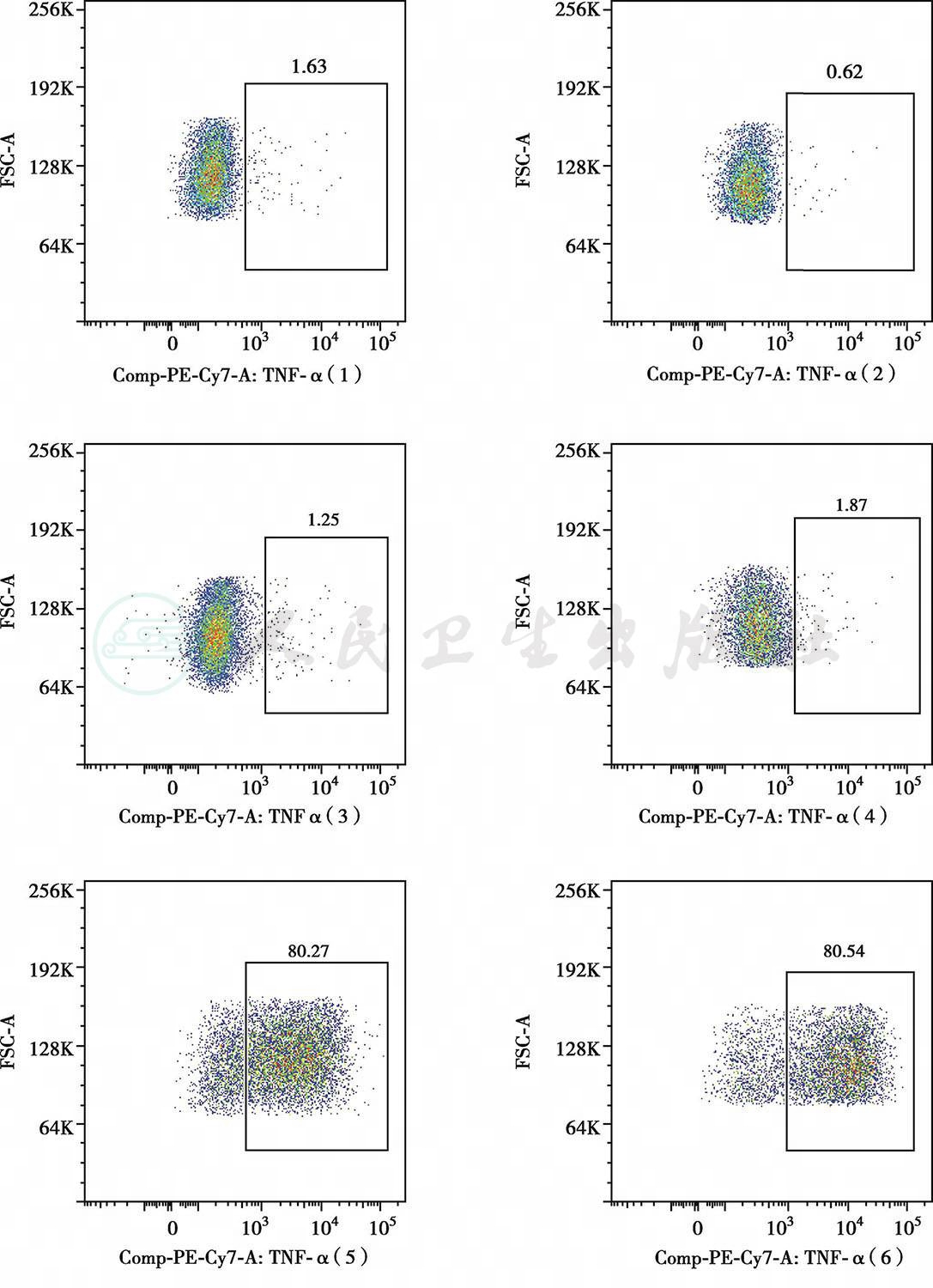
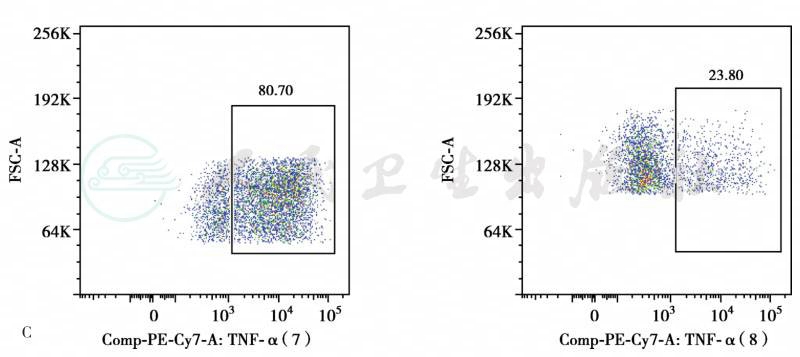
C患儿PBMC在LPS刺激下TNFα产生降低
图1色素失禁症患儿皮疹、疣及TNFα分泌
患儿15岁零11个月,女。出生时面部及全身红血丝。20天时肛周及卡介苗接种处溃烂。4月龄鼻子周围皮肤色素沉着,伴反复发热。皮肤色素沉着逐渐增多。1~2岁开始反复发热、咳嗽、呼吸困难。7岁时因窒息行气管切开,需要反复支气管镜介入治疗局部瘢痕。后反复出现口腔溃疡,伴肛周疙瘩,口唇挛缩影响正常进食。近5年周身反复红斑疹,剧烈瘙痒,与发热有关。近1年出现反复小肠不完全性梗阻。患儿有先天聋哑症,营养不良,中度贫血。WBC 9.26×109/L,N 7.22×109/L,L 1.3×109/L,Hb 76g/L,PLT 538×109/L。CD3 87.5%,CD4 53.6%,CD8 31.5%,B 7.2%,NK 4.7%。IgG 14g/L,IgA 2.75g/L,IgM 1.25g/L,IgE 419IU/ml。胸片示右上及左下肺野条片影,右腋下多发钙化结节影。肺CT示右肺中叶及左肺下叶可见条片状不张,余双肺野散在条索影、片影。双腋下多发增大淋巴结影,右侧淋巴结可见结节样钙化。颈部超声示颈部多发肿大淋巴结。泌尿系统超声示双肾盂小结节样钙化。腹部超声示肝肋下1.8cm。皮肤病理:①(臀部)小块皮肤及皮下组织,表皮过度角化,伴局部角化不全伴中性粒细胞聚集形成小脓肿,棘层增生,真皮浅层局部水肿及纤维素样坏死形成,真皮血管壁部分增厚,血管周及部分附属器周较多淋巴、组织细胞及中性粒细胞,偶见嗜酸性粒细胞浸润。免疫组化:CD20(少量 +),CD30(偶见活化细胞 +),CD3(+),CD68(+),Ki-67(5%+),S-100(色素细胞减少)。原位杂交:EBER(-)。②(下肢)小块皮肤及皮下组织,表皮轻度角化过度,棘层增生,基底细胞小灶空泡变性,真皮血管周散在淋巴、组织细胞、中性粒细胞浸润,可见较多噬色素细胞反应,皮下脂肪组织内亦可见较多淋巴、中性粒细胞及组织细胞浸润。支气管:①送检(支气管)小块组织,镜下大部分为粉染无结构物,其间见脱落的上皮细胞、少量淋巴细胞及浆细胞,大部分细胞退变,其间见革兰氏阳性菌团。②(支气管)涂片见少量的脱落上皮(约50%),淋巴细胞(约40%)及个别单核细胞(约10%),未见肿瘤细胞。特殊染色结果:革兰氏阳性菌团(+),PAS(-),六胺银(-),黏卡(-)。患儿具有杂合新发的IKBKG基因L342P突变。患儿中性粒细胞X染色体为偏移灭活(父/母88:12),淋巴细胞X染色体为轻度偏移灭活(母/父70:30),提示免疫缺陷受累细胞主要为淋巴细胞。患儿单个核细胞脂多糖刺激下肿瘤坏死因子α产生降低
引自:实用儿童原发性免疫缺陷病.第1版.ISBN:978-7-117-32242-3.主编:
NEMO缺陷患儿免疫球蛋白合成紊乱,或高IgM表型,占患儿的23%,但大部分患儿不具有高的IgM,而是明显升高的IgA。相关的突变为累及C417和394的截断蛋白。累及亮氨酸拉链的突变患儿具有正常的免疫球蛋白。大部分患儿对多糖抗原反应受损。NK细胞功能受损。患儿全血细胞对TNF-α刺激后IL-10产生受损。
所有IKBA缺陷患儿具有明显的B细胞缺陷(丙种球蛋白紊乱,记忆的、转换记忆的B细胞降低或完全缺乏,针对疫苗抗原的抗体水平降低或缺乏),一些患儿记忆CD4+T和CD8+T细胞比例降低,TCRγδT细胞缺乏,对anti-CD3的T细胞增殖严重受损。一部分患儿缺乏外周淋巴结,淋巴细胞增多。所有患儿细胞显示针对各种不同表面受体刺激的NF-κB反应受损,包括TLR、IL-2R、TCR和BCR。
预防性治疗包括抗生素预防,如复方新诺明和/或青霉素V的口服。如果患儿表现B细胞免疫受损需要静脉丙种球蛋白替代治疗。如果患儿具有功能性的B细胞免疫,需接种肺炎双球菌结合或非结合疫苗,流感嗜血杆菌结合疫苗,奈瑟脑膜炎球菌结合和非结合疫苗。只要一旦怀疑感染或患儿出现中度发热,马上开始针对肺炎链球菌、金黄色葡萄球菌、铜绿假单胞菌、流感嗜血杆菌的静脉抗生素治疗,不必考虑炎症指标,因为即便已经采取预防措施,患儿可死于迅速的侵袭的细菌感染。下一步的抗生素调整依据病原菌的结果。
世界范围内29例NEMO缺陷患儿行HSCT,24例成功植入,13例具有GVHD。7例在HSCT后0.2~12个月死亡。平均随访57个月(4~108个月),总存活率74%。之前存在的分枝杆菌感染和结肠炎与HSCT预后不良有关。移植不能治愈结肠炎,可能是由于上皮屏障的细胞内在缺陷所致。
7/16例IκBα缺陷患儿死亡(6例行HSCT后),1例1岁前死亡,2例预防下存活(9岁和22岁),11/16例行HSCT,6/11例死于移植中或移植后的细菌败血症、进展性神经退行性变、急性呼吸衰竭、脑出血,5例移植成功,均具有ED表型,4例仍有持续部分免疫缺陷(反复感染、弥漫皮肤疣和慢性腹泻),给予IVIG(HSCT后3~21年)治疗,1例不需要治疗。5例IκBα缺陷患儿移植成功提示可以建议HSCT,但不能校正非血细胞的缺陷,包括细胞自主内在的免疫、一定淋巴器官的发生及外配层发育不良。HSCT后应长期预防感染。不应该接受活疫苗(卡介苗、减毒的脊髓灰质炎疫苗、麻疹疫苗、腮腺炎疫苗、轮状病毒疫苗、风疹疫苗和水痘疫苗)。1例出现播散性卡介苗病。









