


 收藏
收藏 已收藏
已收藏英文名称 :Niemann-Pick disease
中文别名 :Niemann-Pick病
尼曼-皮克病(Niemann-Pick disease,NPD)是由于酸性神经鞘磷脂酶缺乏导致神经鞘磷脂在单核巨噬细胞中堆积所致,为常染色体隐性遗传。
本组疾病为常染色体隐性遗传,目前发现的相关基因有SMPD1、NPC1和NPC2(表1)。
表1 Niemann-Pick病的分型
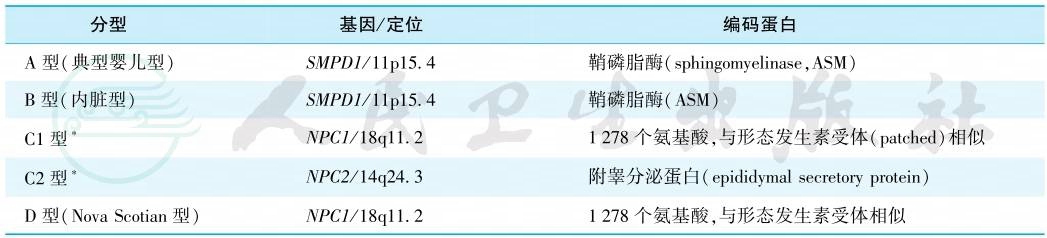
注:∗C型中95%为C1型,5%为C2型。
本病主要病理改变为网状内皮系统丰富的内脏器官中见尼曼-皮克细胞,该细胞直径20~100μm,单个偏心胞核,染色质疏松,可见2~3个核小体,胞质充满空泡,呈泡沫样,PAS染色(periodic acid-Schiff staining)空泡中心呈阴性,泡壁阳性,酸性磷酸酶呈阴性。根据临床症状结合骨髓涂片见尼曼-皮克细胞可初步诊断。确诊依据酶活性检测及基因检测。
1.血常规检查
血细胞数正常或下降;伴脾功能亢进者,常全血细胞数减少。涂片中淋巴细胞、单核细胞胞质中出现特征性空泡是诊断该病的重要实验室诊断依据之一,往往是由于鞘磷脂堆积产生的。
2.骨髓检查
骨髓增生活跃或明显活跃,可见数量不等(往往较多)的形态学上具有特异性的尼曼-皮克细胞,其主要特征是胞质中充满大小较一、透明的脂肪滴,详见图1。涂片中以血膜尾部或两侧边缘多见。典型者通过骨髓检查基本可明确诊断。有的患者伴海蓝组织细胞增多。另外,通过细胞化学染色可辅助细胞的辨认,PAS染色呈阳性(泡壁),SBB染色呈阳性,MPO染色呈阴性。
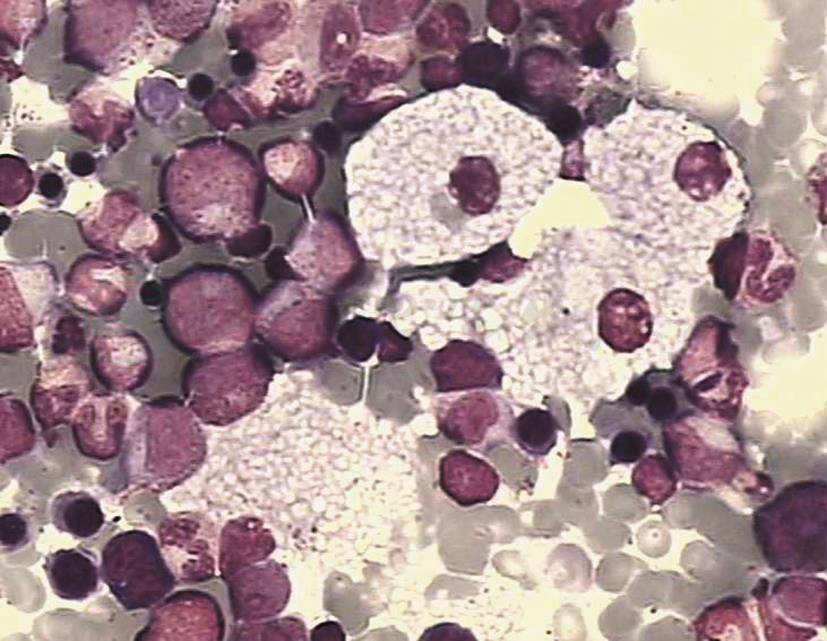
图1 尼曼-皮克病骨髓象(高倍镜)
图中有4个尼曼-皮克细胞,其胞体大,胞质呈泡沫状
在某些疾病的骨髓涂片中也能看到尼曼-皮克样吞噬细胞,如慢性髓细胞白血病、原发免疫性血小板减少症、珠蛋白生成障碍性贫血、GM1神经节苷脂病Ⅰ型、Wolman病、Hurler病、高胆固醇血症、血乳糜微粒过多血症、家族性高密度脂蛋白缺乏症等。两者细胞在形态学上较难区别,但尼曼-皮克样吞噬细胞的胞质中时常还有其他吞噬物,且细胞数量少。
3.组织活检
包括骨髓、肝、脾及淋巴结活检,其切片中可见成堆、成片或弥漫性的泡沫细胞,免疫细胞化学染色CD68呈阳性。故通过组织活检,通常可确诊。
4.酸性鞘磷脂酶活性及细胞内脂质测定
该酶通过人体有核细胞均可提取,临床常从人外周血白细胞或经过培养的皮肤成纤维细胞中提取进行测定,最好与患儿双亲同时测定。A型活性为正常人5%~10%,B型为正常人5%~20%,C型为正常的50% (亦可接近正常或正常),D型及E型酶活性正常。
5.分子生物学检查
采用PCR法,通过基因分型有助于确定临床表型、突变型、了解预后等。
6.细胞超微结构检查
尼曼-皮克细胞在电镜下显示胞质中含有许多颗粒状脂质包涵体,约0. 5~5μm,呈板层状。包涵体在年长的患者中较为明显,年轻或轻症患者则无定形。
从实验室角度来说,根据骨髓检查或组织活检,形态学典型者一般可确断。
1.对症治疗
控制感染,缓解呼吸困难,抗癫痫药控制惊厥,抗胆碱能药物改善肌张力障碍及震颤,输注血小板,镇静及抗惊厥治疗,保证营养供给。
2.酶替代治疗
尚在研究中。
3.底物减少疗法
鞘磷脂合成抑制剂美格鲁特可缓解C型症状,延缓进展。









