


 收藏
收藏 已收藏
已收藏英文名称 :myelodysplastic syndrome
中文别名 :病态造血
骨髓增生异常综合征(myelodysplastic syndrome,MDS)是一组造血干细胞获得性克隆性疾病,常同时或先后出现红细胞、粒细胞和巨核细胞的发育异常(又称病态造血)导致进行性、难治性外周血红细胞、粒细胞及血小板减少;骨髓有无效性造血;临床主要表现为贫血、感染和/或出血。原始细胞增多的MDS转化为AML危险性增高;也有些MDS患者的生物学行为相对惰性,或者向骨髓衰竭方向发展。
原发性MDS病因不明,继发性MDS与接触放射线、苯或接受烷化剂、拓扑异构酶Ⅱ抑制剂类化疗药物治疗有关(于治疗后发生的MDS又称为治疗相关性MDS,2008 年WHO分类归入“治疗相关性髓系肿瘤”)。
MDS的发病机制尚未完全明确。通过大量研究证实MDS是源于骨髓造血干/祖细胞的克隆性疾病。在MDS的发生发展过程中,包含了骨髓细胞的凋亡、增殖及克隆扩张等多重机制。在MDS的患者中细胞遗传学异常较为常见,如5q-、+8、-7等,治疗相关性MDS多为复合染色体异常。表观遗传学的改变在MDS的发病中起重要作用,涉及RNA剪接、DNA甲基化、组蛋白修饰、转录、DNA修复调控、黏结蛋白、RAS通路、DNA复制等多方面的基因突变,造成DNA的高度甲基化和组蛋白去乙酰化等现象。免疫机制异常和骨髓微环境异常在MDS中的发病意义亦得到重视。
有FAB分型和WHO分型。FAB简单,使用方便(表1)。WHO分型取消了难治性贫血名称,将CMML归入MDS/MPN,改变了划分MDS和AML原始细胞标准,结合了细胞遗传学发现,更为全面(表2)。
(一)FAB分型
表1 骨髓增生异常综合征的FAB分型及主要诊断标准
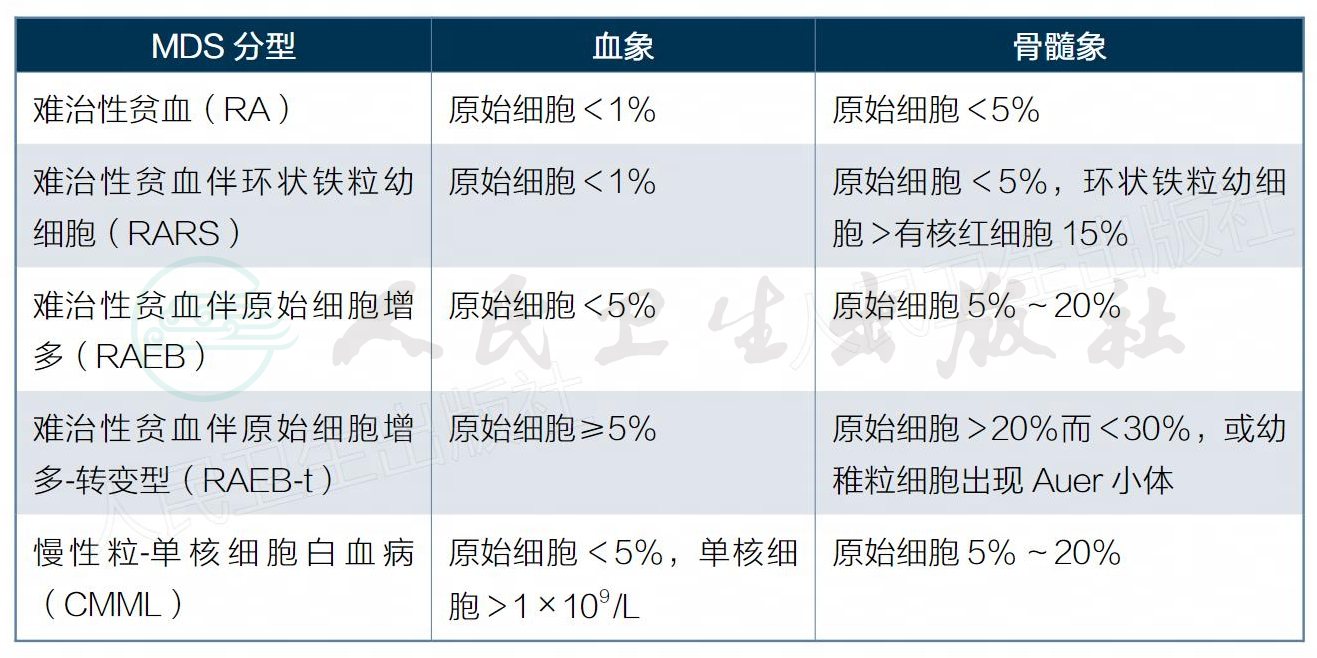
FAB分型中,RAEB及RA所占比例较高,但CMML国内较国外少见。FAB分型有助于判断预后。RA、RAS部分可转变为RAEB、RAEB-t或原始细胞<5%CMML,后者又有部分病例会转变为急性白血病。
(二)WHO分类
FAB分型有许多优点,使用方便,但亦存在不足之处。WHO(2001)造血与淋巴组织肿瘤分类标准结合了细胞遗传学的分析,进一步完善了MDS的诊断分类。2016年又进行了若干补充和修改(表2)。
表2 WHO的MDS分类及其标准

注:*血细胞减少的定义为血红蛋白<100g/L,血小板计数<100×109/L,中性粒细胞绝对计数<1.8×109/L;极少情况下,MDS可见这些水平以上的轻度贫血或血小板减少;外周血单核细胞必须<1×10⁹/L;**如果存在SF3B1突变;***外周血1%的原始细胞必须有两次不同场合检查的记录;△若环形铁粒幼红细胞≥15%的病例有红系明显血细胞发育异常,则归类为MDS-RS-SLD。
(一)血象及骨髓涂片
血象及骨髓象中血细胞形态和数量的异常变化是诊断的重要依据之一。血象中有全血细胞减少的病例占半数以上,部分病例仅为一系或两系血细胞减少。一系减少常为红细胞减少,并可能在全血细胞减少前存在数年。大多数病例骨髓造血细胞呈显著增生。部分病例为增生或增生活跃,仅极少数病例增生低下。增生低下者可称低增生性MDS,仅占10%,但无独立预后意义。90%以上的病例有不同程度的细胞发育异常(病态造血)。
1.红细胞系
骨随中幼红细胞可过多(>60%)或过少(<5%)。红系病态造血主要表现在核的改变,包括核出芽、核间桥、核碎裂、多核和巨幼样变。胞质改变包括特征的环状铁粒幼细胞(含5个以上粗大的铁蛋白颗粒,且围绕核周1/3以上)、空泡及PAS阳性。血象中成熟红细胞大小和形态不一,可有巨大红细胞、椭圆形红细胞及有核红细胞,染色过浅和点彩红细胞。其中以奇数核和巨大红细胞最有诊断意义。
2.粒细胞系
骨髓中原始细胞比例增加,胞体大小不一,核分叶过多或过少,呈假性Pelger-Hüet畸形,胞质嗜碱性强,颗粒可减少或缺乏并分布不均匀,呈假性Chédiak-Higashi颗粒及Auer小体等。血象中有幼稚粒细胞出现和单核细胞增多。其中原始细胞可分为2型:Ⅰ型为无天青颗粒的原始细胞;Ⅱ型为含有天青颗粒但未出现核旁高尔基区的原始细胞。
3.巨核细胞
骨髓中出现淋巴样小巨核细胞、单圆核或多圆核巨核细胞。血片中可有巨大或畸形的血小板。其中以淋巴样小巨核细胞最有诊断意义。
4.细胞化学染色
半数患者骨部分早幼粒细胞和其他不同发育阶段中性粒细胞过氧化酶活性可降低,约20%患者的中性粒细胞碱性磷酸酶积分下降。幼红细胞糖原染色(PAS)可阳性。骨随中除出现环状铁粒幼细胞外,贮存铁亦增加,幼稚粒单细胞α-萘酚醋酸酯酶及氯醋酸酯酶可呈双重阳性。
(二)骨髓活检
通常在骼后上棘取骨髓组织,长度不少于1.5cm。骨髓活检组织中可见到原粒细胞及早幼粒细胞在小梁旁区或小梁间中央区形成集丛(3~5个细胞)或集簇(>5个细胞),称为“幼稚前体细胞异常定位”(abnormal localizationof immature precursors,ALIP),每张骨髓切片上都能看到至少3个集丛或集簇为ALIP(+)。切片中亦可见到幼红细胞聚集成堆,原红细胞增多,伴成熟障碍。小巨核细胞增多。怀疑为MDS的患者建议进行Gomori银染色和原位免疫组化(immuno-histochemical,IHC),常用的检测标志包括CD34、MPO、GPA、CD61、CD42、CD68、CD20和CD3。部分患者伴骨髓网硬蛋白纤维增生。有显著骨髓纤维化约占10%MDS病例,称MDS伴骨髓纤维化,多表示疾病进入进展期,有原始细胞增多。
(三)骨髓染色体检查
常见的变化包括染色体全部或部分缺失,染色体数目增多,但易位少见。常见的染色体变化为-5、5q-、-7、-7q-、+8、20q-和-Y等。5q-综合征常有明显的大细胞性贫血,血小板通常正常或增多,骨髓增生正常或明显增生,巨核细胞的核分叶较少。染色体的变化对随访病情,评估预后有参考意义。在非整倍体的病例中,亚二倍体比超二倍体的预后差。对初诊时核型正常而在随访中出现染色体异常者,预后差,预示有转化为急性白血病的征兆,对有复杂染色体异常者尤为如此。形态学未达到标准(一系或多系细胞发育异常比例<10%)、但同时伴有持续性血细胞减少的患者,如检出具有MDS诊断价值的细胞遗传学异常,应诊断为MDS不能分类(MDS-U)。为提高部分MDS患者细胞遗传学异常检出率,对疑似MDS者,可进行FISH检测,通常探针包括:5q31、CEP7、7q31、CEP8、20q、CEPY和p53等。
(四)流式细胞术检测
目前尚未发现MDS特异性的抗原标志或标志组合,但流式细胞术对于低危MDS与非克隆性血细胞减少症的鉴别诊断有应用价值。对于无典型形态和细胞遗传学证据、无法确诊MDS的患者,流式细胞术检测有辅助诊断参考价值。
(五)分子遗传学检测
对常见基因突变进行检测对于MDS的诊断有潜在的应用价值。随着基因芯片、第二代基因测序等高通量技术的广泛应用,多数MDS患者中可检出体细胞性基因突变,常见突变包括TET2、RUNXI、ASXL1、DNMT3A、EZH2、N-RAS/K-RAS、SF3B1等。部分基因的突变状态对MDS 的鉴别诊断和危险度分层中有一定的价值,推荐作为选做检测项目,包括:TP53、TET2、DNMT3A、IDHI/2、EZH2、ASXL1、SRSF2、RUNX1、U2AF1、SETBP1等。
单核苷酸多态性-微阵列比较基因组杂交技术(SNP-ar-ray)等基因芯片技术可以在多数MDS患者中检测出DNA拷贝数异常和单亲二倍体,进一步提高MDS患者细胞遗传学异常的检出率,在有条件的单位可作为常规核型分析的有益补充。
MDS患者自然病程和预后的差异性很大,应根据病情的不同进行个体化的治疗。应根据MDS患者的预后分组,同时结合患者年龄、体能状况、合并疾病、治疗依从性等进行综合分析,选择治疗方案。较低危组MDS患者的治疗目标是改善造血、提高生活质量,较高危组MDS治疗目标是延缓疾病进展、延长生存期和治愈
(一)对症支持治疗
对一些病情较稳定低危患者,以支持疗法为主,同时密切观察病情及血象的变化。对大多数有较严重贫血(Hb<60g/L)的患者,可以输红细胞以改善贫血。TGF-β抑制剂可适当改善部分低危MDS患者存在的输血依赖。对于红细胞输注依赖的患者应定期监测血清铁蛋白(SF)水平、累计输血量和器官功能(心、肝、胰腺),评价铁超负荷程度。对严重血小板减少(<10×109/L)伴有明显出血倾向时输注单采血小板悬液。有感染时应给予抗生素治疗,控制感染。
(二)促造血治疗
1.雄激素
雄激素对部分有贫血表现的MDS患者有促进红系造血作用,是MDS治疗的常用辅助治疗药物,包括达那唑、司坦唑醇和十一酸睾酮。接受雄激素治疗的患者应定期检测肝功能。
2.红细胞生成素(EPO)
推荐40000~60000IU/周,分次注射,至少使用8周评估。对于轻度贫血的RA、RARS患者且血清EPO水平较低,体外造血细胞培养CFU-E,BFU-E对EPO有反应者效果较好,血清EPO水平>500U/L者疗效较差。
3.粒细胞-巨噬细胞集落刺激因子(GM-CSF)和粒细胞集落刺激因子(G-CSF)
不推荐长期单独使用,仅在反复或伴有耐药感染的粒细胞缺乏患者中可以使用剂量60~200μg/(m²·d),皮下注射,疗程视病情需要确定,一般2~8周。对于原始细胞过多的RAEB患者应在化疗的基础上使用。
4.促血小板生成素(TPO)
剂量1μg/(kg·d),皮下注射,主要用于血小板有明显减少者,7~14天。
5.白细胞介素-11(IL-11)
用于血小板减少的治疗,25~50μg/(kg·d),皮下注射,7~14天。
6.艾曲泊帕
起始剂量和最大剂量可以高于治疗ITP用量。
(三)去甲基化药物
常用的去甲基化药物包括5-阿扎胞苷(azacitidine,AZA)和5-阿扎-2-脱氧胞苷(decitahine,地西他滨)。去甲基化药物可应用于较高危组MDS患者,与支持治疗组相比,去甲基化药物治疗组可降低患者向AML进展的风险、改善生存。较低危组MDS患者如出现严重粒细胞减少和/或血小板减少,也可应用去甲基化药物治疗,以改善血细胞减少。
1.阿扎胞苷
推荐用法为75mg/(m²·d)×7天,皮下注射,28天为1个疗程。接受AZA治疗的MDS患者,首次获得治疗反应的中位时间为3个疗程,约90%治疗有效的患者在6个疗程内获得治疗反应。因此,推荐MDS患者接受AZA治疗6个疗程后评价治疗反应,有效患者可持续使用。高龄、血象较低、骨髓增生低下者耐受性也相对比较好。
2.地西他滨
最佳给药方案仍在不断探索中,较低危组MDS患者地西他滨最佳给药方案迄今尚未达成共识。推荐方案之一为20mg/(m²·d)×5天,每4周为1个疗程。推荐MDS患者接受地西他滨治疗4~6个疗程后评价治疗反应,有效患者可持续使用。部分患者骨髓抑制较明显。
(四)免疫调节剂治疗
沙利度胺:50~200mg,每晚1次。部分患者可改善红系造血,减轻或脱离输血依赖;便秘、周围神经炎等不良反应常见。
来那度胺,其抗肿瘤和免疫调节作用比沙利度胺更强,而不良反应明显低于沙利度胺,推荐剂量为10mg/d,连用21天,4周为1个疗程。对于伴有del(5q)±1种其他异常(除-7/7q-外)的较低危组MDS患者,如存在输血依赖性贫血,可应用来那度胺治疗,部分患者可减轻或脱离输血依赖,并获得细胞遗传学缓解,延长生存。对于不伴有del(5q)的较低危组MDS患者,如存在输血依赖性贫血,且对细胞因子治疗效果不佳或不适合采用细胞因子治疗,也可以选择来那度胺治疗。伴有5q-的MDS患者,若出现下列情况不建议使用来那度胺:骨髓原始细胞比例>5%;复杂染色体异常;IPSS-中危2或高危组;检出p53基因突变。
(五)化疗
1.小剂量化疗
常采用小剂量阿糖胞苷(Ara-C)10~20mg/(m²·d),14~21天为1个疗程;三尖杉酯碱(H)1mg/d,10~14天为1个疗程;阿克拉霉素3~14mg/(m²·d),7~10天为1个疗程;或依托泊苷(VP16),25~35mg/(m²·d),7~10天为1个疗程。预激方案为在小剂量阿糖胞苷(10mg/m²,每12小时1次,皮下注射,14天)基础上加用G-CSF,并联合阿克拉霉素或高三尖杉酯碱或去甲氧柔红霉素,常用于老年或身体机能较差的相对高危组患者;预激方案可联合去甲基化药物使用。
2.联合化疗
按照AML标准方案治疗。
(六)免疫抑制剂治疗
抗胸腺淋巴细胞球蛋自(ATG)与环孢素A,通过抑制CD8细胞来调节MDS的免疫反应。ATG(马抗)剂量15~40mg/(kg·d),连用4~5天;环孢素A剂量3~5mg/(kg·d),3~6个月。ATG和环孢素可以合用,用于年龄≤60岁、属较低危组,伴骨髓原始细胞比例<5%、骨髓增生低下、正常核型或单纯+8、存在输血依赖、HLA-DR15阳性、存在PNH克隆或携带有STAT-3突变细胞毒T细胞克隆的患者。原始细胞>5%、-7或有复杂染色体等情况下不宜使用。
(七)造血干细胞移植
异基因骨髓移植(allo-HSCT)是目前可能治愈本病的一种治疗方法,其适应证:①年龄<65岁、较高危组MDS患者;②年龄<65岁、伴有严重血细胞减少、经其他治疗无效或伴有不良预后遗传学异常(如-7、3q26重排、TP53基因突变、复杂核型、单体核型)的较低危组患者。近年,减低强度的预处理方案受到重视,该法降低移植相关死亡率,而疗效基本不受影响。拟行allo-HSCT的患者,如骨髓原始细胞≥5%,在等待移植的过程中可应用化疗或联合去甲基化药物桥接allo-HSCT。
MDS国际工作组(International Working Group,IWG)于2000年提出国际统一疗效标准,2006年又进一步修订,使不同临床治疗方案结果间具有可比性。MDS的治疗反应包括以下四种类型:改变疾病的自然病程、细胞遗传学反应、血液学改善和改善生存质量。









