


 收藏
收藏 已收藏
已收藏英文名称 :hemoglobinopathy
血红蛋白病(hemoglobinopathy)是由于血红蛋白一级分子结构异常(异常血红蛋白病),或由于一种或一种以上珠蛋白肽链不能合成或合成不足,但缺失或不足的珠蛋白肽链一级分子结构正常(珠蛋白生成障碍性贫血,原称地中海贫血及海洋性贫血)所引起的一组遗传性血液病。临床可表现为溶血性贫血、高铁血红蛋白血症或因血红蛋白氧亲和力增高或减低而引起组织缺氧或代偿性红细胞增多所致发绀。
血红蛋白病种类繁多,临床症状多样化,但归纳其结构变异所导致功能异常,大致分为以下数类:
1.因分子内部氨基酸替代所产生的异常血红蛋白血红蛋白分子内部为非极性氨基酸,如被不同理化性质的氨基酸替代,会影响分子的构型和稳定性,此类异常血红蛋白包括血红蛋白M(Hb M)、不稳定血红蛋白(UHb)和氧亲和力改变的血红蛋白。
(1)Hb M:肽链中与血红素铁原子连接的组氨酸被酪氨酸所替代,最常见的是E7或F8的组氨酸为酪氨酸所替代,酪氨酸酚基上的氧与血红素的铁原子构成离子键,使铁原子呈稳定的高铁状态,影响血红蛋白的正常释氧功能,使组织供氧不足,出现发绀及红细胞增多。高铁血红素并易与珠蛋白链分离,使血红蛋白分子结构不稳定而发生溶血。
(2)UHb:肽链中与血红素紧密结合的氨基酸发生替代或缺失,影响肽链的立体结构或减弱与血红素的结合力,形成UHb分子。水易进入血红蛋白腔内,使亚铁血红素氧化为高铁血红素;β链第93位半胱氨酸的硫氢基被氧化,产生硫化物,形成硫化血红蛋白,使珠蛋白链与血红素分离。游离珠蛋白链在37℃即不稳定,四聚体易解离为单体,在红细胞内聚集沉淀,形成包涵体,使细胞膜僵硬,通过微循环时往往导致膜部分丧失,最终变为球形红细胞,在脾阻留而破坏。
2.因分子外部氨基酸替代所产生的异常血红蛋白种类很多,一般均对分子构型、功能和稳定性没有明显影响。
(1)Hb E是β链第26位谷氨酸被赖氨酸替代,因谷、赖两种氨基酸理化性质相同,其替代位置虽在α1β1接触面上,但对血红蛋白分子的稳定性和功能影响不大。这类异常血红蛋白中少数可产生溶解度改变。
(2)Hb S和Hb C均由于其分子外部形状或电荷改变,缺氧时溶解度降低。Hb S聚合为螺旋状体,扭曲成镰刀形纤维;而Hb C聚合成一种副结晶,两者均使细胞膜变硬,难以通过微循环,丧失部分红细胞膜,形成球形红细胞,在脾窦内阻留溶破。
(3)β珠蛋白生成障碍性贫血患者,过剩的α肽链形成多聚体,引起红细胞膜损害,致使大量幼红细胞无效生成。
(4)α珠蛋白生成障碍性贫血,过剩的β及γ链形成Hb H(β4)或Hb Bart's(γ4)。Hb H是一种不稳定血红蛋白,Hb H包涵体结合在红细胞膜上,使膜对阳离子通透性发生改变,钾盐与水逐渐从红细胞内渗至胞外。缺钾红细胞寿命缩短,易在单核-巨噬细胞系统被破坏,导致溶血。Hb Barts对氧亲和力增高,造成组织缺氧。
人类血红蛋白是一种结合蛋白,由珠蛋白和亚铁血红素组成,分子量64400,血红素由原卟啉与亚铁原子组成。每一个血红蛋白有两对珠蛋白肽链,一对是α链,包括α与ζ2种肽链,由141个氨基酸残基构成,含较多组氨酸,在运氧中具有重要生理作用。另一对是非α链,有β、γ、δ及ε4种肽链。ζ、ε、α与γ链,分别组成胚胎早期(妊娠3个月以内)血红蛋白、Hb Gower1(ζ2ε2)、Hb Gower2(α2ε2)、Hb Portland(ζ2γ2)。β链含146个氨基酸残基,β93半胱氨酸易被氧化产生混合二硫化物及其他硫醚类物质,可降低血红蛋白稳定性。δ链亦由146个氨基酸残基组成,仅10个氨基酸与β链不同。由于δ链中第22位丙氨酸置换了β22谷氨酸,第116位精氨酸换了β116组氨酸,因此δ链的正电荷大于β链,Hb A2(α2δ2)等电点升高,电泳时靠近负极。γ链虽由146个氨基酸组成,但与β链有39个氨基酸不同,且含有4个异亮氨酸,为α、β与δ链所缺如,因此可用分析异亮氨酸方法测定Hb F(α2γ2)含量。初生时Gγ与Aγ的比例是3∶1,儿童和成人两者之比为2∶3。每一条肽链和一个血红素连接,构成一个血红蛋白单体,人类血红蛋白是由两对(4条)血红蛋白单体聚合而成的四聚体。人类血红蛋白中珠蛋白结构略有不同,但血红素相同。
血红蛋白的四级结构:由氨基酸顺序排列的肽链结构称为血红蛋白的一级结构。肽链中的氨基酸可分为亲水的极化氨基酸(其侧链为羧基、氨基),与非极化的氨基酸(其侧链是芳香族)。肽链中的各种氨基酸的侧链相互拉紧形成α螺旋,螺旋形节段间由短而非螺旋形节段相连。螺旋形节段从N端至C端分别以A-H表示(图1),α类肽链包含7个螺旋(无D螺旋),非α类肽链包含8个螺旋片段。非螺旋形节段用AB、CD等表示,称为血红蛋白二级结构。血红素的铁原子有6个配位键,第5个配位键结合在肽链F段第8位氨基酸上,第6个配位键结合氧,并间接结合在肽链E段的第7位氨基酸上。肽链围绕血红素为中心,构成内外两层螺旋状蛇形盘曲的三维空间结构,称为三级结构(图16-2-16)。亲水氨基酸分布于外层,使血红蛋白能溶于水而不致沉淀;疏水氨基酸分布于内层,使水分子不能进入血红素腔内部,避免血红素的Fe2+氧化为Fe3+。四个血红蛋白单体(肽链三级结构加血红素),按一定的空间关系结合成四聚体,如Hb A(或Hb A 1,α2β2)、Hb A 2(α2δ2)及Hb F(α2γ2),称异质型四聚体;由两对同样的三级结构血红蛋白单体结合成的四聚体,如Hb H(β4)及Hb Bart(γ4),称为同质型四聚体。以上所述四聚体为血红蛋白四级结构。综上所述,血红蛋白与分子的外表结构必需完整,带有负电荷,α、β链结合部位要固定,包围血红素腔的氨基酸顺序排列需完整,否则血红蛋白就不能维持分子结构稳定性及正常运氧生理功能,并易遭破坏。
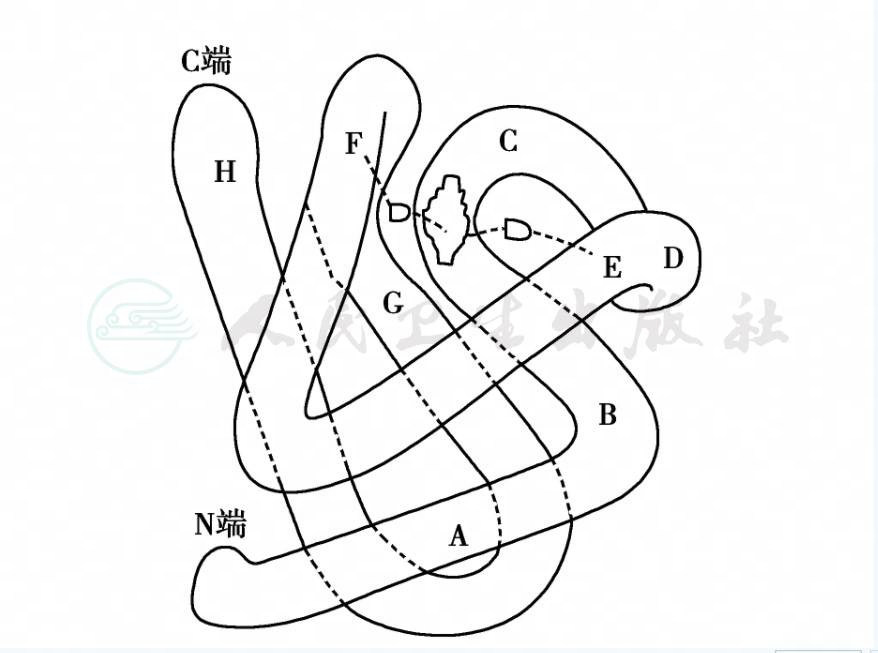
图1 血红蛋白三级结构示意
肽链N~C端折叠成8个螺旋节段(A~H),螺旋节段由非螺旋节段(AB~GH)相连,血红素位于中心与F8 E7组氨酸相连,构成内外两层螺旋状蛇形盘曲的三维空间结构
正常人出生后有三种血红蛋白:①血红蛋白A(Hb A),由一对α链和一对β链组成(α2β2),占正常成人及6岁以上儿童血红蛋白总量的90%以上。胚胎2个月时Hb A即有少量出现,初生时占10%~40%,出生6个月后即达成人水平。②血红蛋白A2(Hb A 2),由一对α链和一对δ链组成(α2δ2)。自出生6~12个月起,占血红蛋白的2%~3%。③胎儿血红蛋白(HbF),由一对α链和γ链组成(α2γ2),初生时占体内血红蛋白的70%~90%,以后渐减直至生后6个月其含量降至血红蛋白总量的1%左右。血红蛋白的不同肽链是由不同的遗传基因控制的,α链基因位于第16号染色体短臂,β、δ、γ链位于第11号染色体短臂,呈连锁关系。
珠蛋白基因突变而致肽链的单个或多个氨基酸替代或缺如,导致珠蛋白分子结构改变,称为异常血红蛋白。若α珠蛋白基因的缺失或缺陷,可导致α珠蛋白链合成减少或缺乏,称为α珠蛋白生成障碍性贫血。若β珠蛋白基因缺陷,可导致β珠蛋白链合成减少或缺乏,称为β珠蛋白生成障碍性贫血。全世界范围内经结构分析证实的异常血红蛋白日益增多,但不到1/3的异常血红蛋白会产生临床症状。世界卫生组织估计,全球约有1.5亿人携带血红蛋白病基因,并已将血红蛋白病列为严重危害人类健康的6种常见疾病之一。主要集中在热带和亚热带地区,好发于地中海沿岸、美国黑人人群、北非、东南亚和印度次大陆等地区,世界上有4.83%的人口携带珠蛋白变异基因。在我国以广西、广东、海南等省发病率较高,其中广西人群中地中海贫血基因携带率为12.22%~23.02%。
血红蛋白病的分子遗传学变化大致可归纳为以下数类:
1.单个碱基替代
由于遗传密码中单个碱基替代,导致由该碱基决定的氨基酸发生相应的变化,形成肽链中单个氨基酸置换的异常血红蛋白,例如Hb S、Hb C等。目前发现的异常血红蛋白中,以本类型最多见,约占90%。
2.终止密码的突变
因终止密码的变异使珠蛋白肽链不在正常的位置终止,导致肽链延长或缩短,如Hb Mc-KeesRock的β链第145位氨基酸的碱基由UAU变为UAA(终止密码),使β链提前结束,仅含144个氨基酸。
3.移码突变
如正常血红蛋白肽链遗传密码中,嵌入或缺失1~2个碱基,使正常三联密码子碱基成分发生改变,如Hb Tak为β链第147位终止密码UAA前插入AC,使UAA→Thr(苏氨酸),而致β链延长至第157位氨基酸,比正常β链多11个氨基酸。
4.密码子缺失或插入
生殖细胞减数分裂时,染色体发生错配或不等交换,形成两种珠蛋白基因。一种失去一部分密码子,合成缺失部分氨基酸的肽链,如Hb Lyon的β链第17~18位缺失赖氨酸、缬氨酸,另一条染色体单体上却嵌入了相应密码子,合成插入部分氨基酸的肽链。β珠蛋白基因和δ珠蛋白基因大片段缺失,可致遗传性胎儿血红蛋白持续存在综合征,或δβ珠蛋白生成障碍性贫血。
5.融合基因
减数分裂时,不同珠蛋白基因之间发生不等交换,合成融合链的异常血红蛋白,如δ链和β链基因错误联合,产生不等交换,形成融合δβ(Hb Lepore)和βδ(Hb反Lepore)。
6.启动子突变
启动子区有单个核苷酸被置换,使启动子功能降低。如β珠蛋白基因启动子在内的小片段缺失,可导致β珠蛋白生成障碍性贫血,表现为Hb A 2显著升高,可能因主要的转录因子和基因座控制区(LCR)在β珠蛋白基因启动子缺失情况下与δ珠蛋白基因启动子相互协同作用所致。
7.剪接突变
β珠蛋白基因第24~27位密码子突变易发生剪接位点激活,如β24位点GGT→GGA突变(均编码甘氨酸)不引起氨基酸替代,但可改变剪接过程,导致β珠蛋白生成障碍性贫血。天然剪接位点或内含子中的重要位点发生点突变会减少或抑制剪接加工,导致珠蛋白生成障碍性贫血。
在此病高发地区及患者家系中,务必行婚前检查、遗传咨询及血红蛋白病筛查工作,宣传近亲结婚的危害性,劝阻双方均为本病基因携带者婚配;对高危家系应作产前诊断,早期发现重型胎儿,劝其终止妊娠。对于高发地区已应用RFLP连锁分析,间接检测β珠蛋白生成障碍性贫血基因,或应用人工合成的寡核苷酸探针进行杂交,直接检测突变基因等基因诊断技术。









