


 收藏
收藏 已收藏
已收藏英文名称 :glucose-6-phosphate dehydrogenase deficiency
葡萄糖-6-磷酸脱氢酶缺乏症(glucose-6-phosphate dehydrogenase deficiency)是最常见的红细胞酶病。
G6PD基因定位于X染色体(Xq28)。遗传方式为X伴性不完全显性遗传,具有不同的表现度。男性患者为半合子,由于只有一条X染色体,故酶活力显著缺乏,男性患者与正常女性婚配,所生儿子全部正常,女儿全部为杂合子。女性有两条X染色体,女性杂合子的另一条X染色体等位基因正常,通常溶血代偿良好,而无贫血,但如酶活力显著减低时也可有临床症状。女性杂合子与正常男性婚配,有50%概率遗传给后代,获得突变基因的儿子有临床表现,女儿则50%可能为杂合子。女性纯合子必须父母均有缺陷,可有严重溶血表现,女性纯合子与正常男性婚配,儿子携带该缺陷基因的半合子,女儿均为杂合子。基因突变影响G6PD的编码,迄今已报告180多种基因突变,大多涉及错义突变,单个氨基酸被置换。中国人G6PD基因突变类型与国外报道有显著区别,我国最常见的突变型为G1376T、G1388A和A95G。
几乎所有的磷酸戊糖旁路缺陷均是因葡萄糖-6-磷酸脱氢酶(glucose-6-phosphate dehydrogenase,G6PD)缺乏所致,这是和溶血性贫血相关的最常见的酶异常,全世界约有4亿人受累。红细胞G6PD遗传缺陷患者遍及世界各大洲,以东半球的热带和亚热带地区为主,不同种族的发生率有很大差异,最高者为土耳其东南部的犹太人(58.2%),也多见于美国及非洲黑种人、意大利和希腊白种人,以及西班牙和葡萄牙血统犹太人。因为疟疾流行区G6PD缺陷发生率特别高,所以认为G6PD缺陷可能是逃避恶性疟疾感染的一种优势选择。我国广西壮族自治区的某些地区(15.7%)、海南省黎族(13.7%)、云南省傣族发病率较高,其次为四川省简阳市及广东省等。复旦大学附属华山医院采用荧光斑点试验普查发现,上海地区一般人群G6PD缺乏症患病率为0.87%(标化率1.38%)。
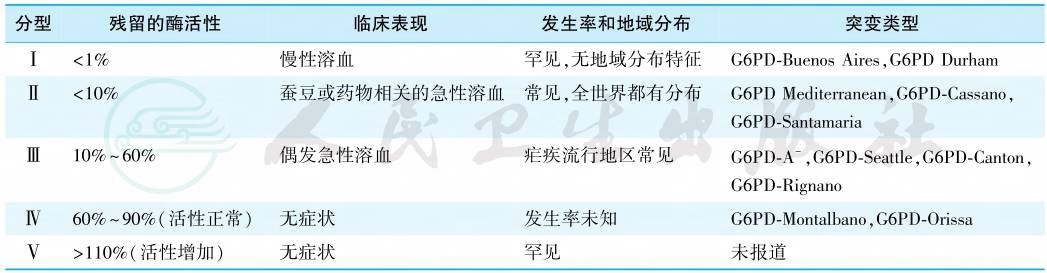
虽然已报道有180种以上G6PD基因突变型,导致400种以上的生化变异型,但常见的酶变异型只有少数几种,例如G6PD A-(GdA-)、G6PD Mediterranean(GdMed)、G6PD Canton(GdCanton)、G6PD Seattle、G6PD Union 变异型等,其中 GdA-占绝大多数。WHO根据G6PD缺乏程度和溶血严重度将G6PD变异型分为5型(表1)。Ⅰ型有严重的酶缺陷引起慢性非球形红细胞性溶血性贫血;Ⅱ型也有严重的酶缺陷,通常在蚕豆或药物作用下会出现间歇性的急性溶血;Ⅲ型中度酶缺陷,偶发急性溶血;Ⅳ型酶活力正常;Ⅴ型酶活力反而增高。Ⅳ型和Ⅴ型无临床意义。
表1 G6PD缺乏症的WHO分型
G6PD缺陷所致的溶血性贫血的诊断除服药史、家族史和临床表现外,主要依靠实验室检查,其方法有以下几种。
(一)筛查试验
1.荧光斑点试验
如果受检标本中G6PD活性正常,则能将试剂中的NADP还原为NADPH,后者在长波紫外线(260~340nm波长)的照射下,发出蓝色荧光。10分钟内出现荧光为正常,10~30分钟间出现为中间缺乏值,30分钟不出现为严重缺乏。如果G6PD活性低于25%即无荧光产生。本试验操作方便,筛检试验中特异性最高。
2.硝基四氮唑蓝纸片法
还原型辅酶Ⅱ(NADPH)通过吩嗪二甲酯硫酸盐的递氢作用,使硝基四氮唑蓝(淡黄色)还原成紫色的甲䓬。NADPH生成的量与甲䓬产生的量在一定范围内呈线性关系。根据颜色变化,判断有无G6PD缺乏。正常酶活性者,温育后纸片应转为紫色。酶活性缺乏者,纸片仍为红色。酶活性中间值或女性杂合子,纸片颜色介于正常与缺乏中间,为淡紫色。
3.高铁血红蛋白还原试验
以高铁血红蛋白还原率间接反映磷酸戊糖旁路代谢状态。在G6PD活性正常者还原率为≥75%,中度缺乏者为31%~74%,重度缺乏者≤30%。该试验简便,敏感性较高,但假阳性高,逐渐被淘汰。
4.红细胞Heinz小体计数
正常红细胞中不应发现Heinz小体。凡能引起高铁血红蛋白的化学物几乎都能在红细胞内产生Heinz小体,G6PD缺乏及不稳定血红蛋白病导致溶血时也可发现Heinz小体。G6PD缺乏导致急性溶血后48小时内,Heinz小体明显增多。红细胞内Heinz小体>5%,有诊断意义。
(二)确诊试验
1.G6PD酶活性定量测定
催化特异底物的酶活力定量法是诊断的“金标准”。国际血液学标准化委员会(ICSH)推荐Beulter确立的速率法,通过紫外分光光度计监测37℃条件下红细胞G6PD酶反应初速度阶段催化产生的产物NADPH含量来计算酶活力。该方法是基于WHO推荐的Zinkhamf法、Glock法、Mclean法的改良。G6PD活力正常参考值:(12.10±2.00)EU/gHb(37℃);(8.34±1.95)EU/gHb(37℃,G6PD校正)。
2.基因型鉴定
应用PCR方法进行已知突变基因的鉴定。最常见的有G6PD A-、地中海G6PD Mediterranean和亚洲G6PD Canton。
上述各项试验必须在溶血高峰时操作,必要时2~3个月重复检验。同时检查患者母亲更有意义。此外,尚须注意和获得性G6PD缺乏症鉴别,白血病和MDS可有多种红细胞酶活性改变。复旦大学附属华山医院统计获得性G6PD缺乏症的患病率高达43.1%(标化率42.8%)。
G6PD缺乏症往往有家族史、无法解释的新生儿高胆红素血症、外周血出现咬痕细胞和Heinz小体、在感染或服用药物或食用蚕豆后急性发作,这些均有助于临床诊断,确诊需要进行上述实验室检查。荧光斑点筛选试验和直接测定G6PD活力为最常用的试验。
在没有外源性氧化剂作用的情况下,绝大多数G6PD缺陷者的红细胞表现正常,因此G6PD缺陷本身不需要治疗。防治要点是避免氧化剂的摄入和积极控制感染。轻中度急性溶血者需立即停服相关药物或控制相应的感染,严重溶血者需少量反复输血。由于G6PD缺乏引起的新生儿溶血与一般新生儿溶血的处理基本相同,为了防止神经系统受损,当未结合胆红素>150μmol/L时需要光疗,>300μmol/L时需要输注红细胞进行换血疗法。注意水电解质平衡并保持足够多的尿量,警惕肾衰竭的发生。应用有关药物前,均应询问患者及其家属有无溶血或红细胞G6PD缺陷病史。抗氧化剂(维生素E、硒)疗效不肯定。不推荐切脾治疗,除非是依赖输血的严重病例。









