


 收藏
收藏 已收藏
已收藏英文名称 :Fanconi syndrome
范科尼综合征(Fanconi syndrome,也称为Fanconi综合征)是由于遗传性或获得性疾病导致近端肾小管整体功能异常引起的一组症候群,因肾脏过多丢失而产生全氨基酸尿、葡萄糖尿、磷酸盐尿、碳酸氢盐尿以及尿酸等有机酸尿;由于过多丢失电解质而产生各种代谢性并发症,如高氯性代谢性酸中毒、脱水、电解质紊乱、佝偻病、骨软化症、生长迟缓等。
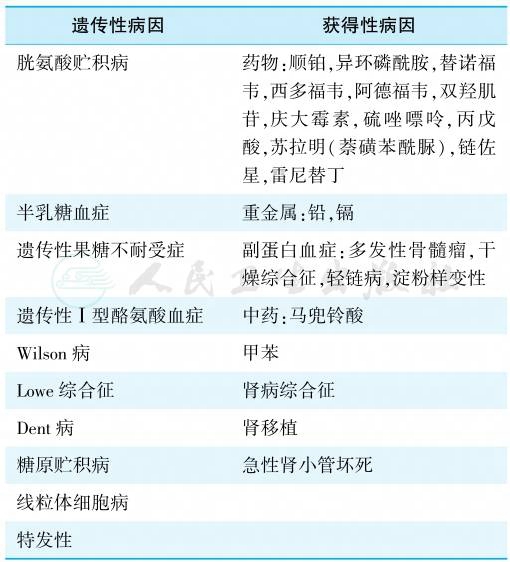
Fanconi综合征的发病机制尚未完全阐明,且因不同病因而异。可能的机制包括:近端小管载体的广泛异常、刷状缘或基底侧细胞膜“渗漏”、Na+-K+-ATP酶抑制或功能异常、线粒体能量代谢异常或其他细胞器功能障碍。儿童Fanconi综合征最常见的病因是先天性转运和代谢异常,而成人Fanconi综合征最常见的病因则是内源性或外源性毒素。具体的病因分类见表1。
表1 Fanconi综合征的病因
1.氨基酸尿
全氨基酸尿是Fanconi综合征的主要特征。由于相对于饮食摄入而言,尿液中损失的氨基酸微不足道,为0.5~1.0g/d,因此没有临床后果。
2.糖尿
糖尿是Fanconi综合征的另一个主要特征,因肾小管对葡萄糖的重吸收受损所致,通常是最早的诊断线索之一。与氨基酸尿一样,糖尿很少引起诸如体重减轻或血糖过低等症状。
3.低磷血症
由于肾脏对磷酸盐重吸收损害所致的低磷(酸盐)血症也是Fanconi综合征常见的临床表现。血PTH水平升高和维生素D含量降低也可能参与了低磷血症的发病;低磷血症的另一种机制是megalin依赖性PTH降解受损。低磷血症通常会导致严重的骨病,并伴有疼痛、骨折、佝偻病或生长发育迟缓。
4.高氯性代谢性酸中毒
是近端肾小管碳酸氢根重吸收障碍所致(近端或2型肾小管酸中毒),正常滤过碳酸氢根的重吸收下降超过30%,而血清碳酸氢根离子浓度通常保持在12~18mmol/L。
5.肾脏丢失钠、钾
Fanconi综合征的患者在碳酸氢根丢失的同时常伴有钠、钾的丢失,严重时甚至可危及生命。
6.多尿、多饮
多尿、多饮和频发严重脱水是年轻的Fanconi综合征患者的常见症状。
7.发育迟缓
Fanconi综合征儿童的发育迟缓是多方面的。低磷血症、佝偻病和酸中毒会导致发育迟缓,慢性低钾血症和细胞外液容量收缩也会加重发育障碍。
8.蛋白尿
除非Fanconi综合征合并肾病综合征,通常蛋白尿很少,且以低分子量蛋白尿(<30 000)为主。
1.胱氨酸贮积症
又称胱氨酸增多症,是小儿Fanconi综合征最常见的病因,其生化特征是胱氨酸在细胞内尤其是在溶酶体中贮积过多。
本病是一种常染色体隐性遗传疾病,由CTNS基因突变引起。往往在出生6个月后首先出现Fanconi综合征的临床症状和体征,出生1年后表现为佝偻病和发育迟缓,肾小球滤过率(GFR)持续下降,一般尚未进入青少年期就进展为终末期肾病。常见晚期并发症包括甲状腺功能减退、肝脾大、视力下降、吞咽困难、肺功能不全和角膜溃疡。老年患者可有血管尤其是冠状动脉的钙化,进而导致心肌缺血。
本病诊断的依据是细胞内胱氨酸水平升高,角膜裂隙灯检查发现胱氨酸结晶强烈提示本病的诊断。羊水或绒毛膜标本直接检测胱氨酸含量对产前诊断有很大帮助。
本病的非特异性疗法包括补充维生素D和纠正酸中毒及电解质紊乱,终末期肾衰竭时需替代治疗。半胱胺疗法可降低组织中的胱氨酸水平并延缓GFR的降低,尽早开始治疗(在2岁之前)可改善发育迟缓,但不能改善Fanconi综合征。
2.半乳糖血症
为半乳糖代谢异常的常染色体隐性遗传疾病,由半乳糖1-磷酸尿嘧啶转移酶的活性降低引起。当患儿摄入含半乳糖的牛奶后,会迅速出现呕吐、腹泻,以后出现黄疸、肝大、白内障、智力发育迟缓和Fanconi综合征。治疗主要为无半乳糖饮食。
3.遗传性果糖不耐受症
本病是另一种与Fanconi综合征有关的影响碳水化合物代谢的常染色体隐性遗传疾病。因果糖1-磷酸醛缩酶B异构体缺乏,导致1-磷酸果糖不能裂解而储积在细胞中,同时不能产生ATP而影响细胞能量代谢,出现近端小管功能障碍和乳酸酸中毒。患儿在摄入果糖后不久可发生急性Fanconi综合征,出现恶心,呕吐和低血糖症状,甚至出现惊厥、休克和急性肾损伤。治疗为严格限制果糖、蔗糖和山梨醇饮食。
4.糖原贮积症(glycogen storage disease,GSD)
大多数为常染色体隐性遗传病,其特征为糖尿过多以及肝脏和肾脏中糖原大量贮积。本病主要是由于肾小管和肠上皮细胞葡萄糖转运蛋白2(glucose transporter 2,GLUT2)的活性不足,无法将葡萄糖从近端小管和肠上皮细胞的基底侧运出,另外也使葡萄糖无法进出肝细胞和胰腺β细胞。该疾病的治疗主要针对肾脏溶质流失,治疗佝偻病以及频繁进食以预防酮症。
5.酪氨酸血症
是罕见的常染色体隐性遗传病。由于富马酰乙酰乙酸盐水解酶、马来酰乙酰乙酸盐水解酶缺陷,导致琥珀酰丙酮升高,引起Fanconi综合征。肾脏的损伤则是琥珀酰丙酮的毒性作用所致。急性型出生早期就出现肝功能失代偿;慢性型变现为Fanconi综合征、肝硬化和低血磷佝偻病、肝癌。早期予尼替西农,90%患者有治疗反应。晚期肝硬化患者可行肝移植术。
6.Wilson病
Wilson病是影响铜代谢的常染色体隐性遗传疾病,是由P型铜转运腺苷三磷酸酶ATP7B的缺陷引起的,影响肝脏、肾脏和中枢神经系统,导致组织中大量铜的贮积。Fanconi综合征通常出现在肝衰竭之前,还可出现高钙尿症,近端和远端小管功能均有障碍。根据疾病严重程度,给予1.0~1.5g/d的青霉胺治疗可逆转肾功能不全,并可逆转肝脏和神经系统病变。
7.Lowe综合征
又称眼-脑-肾综合征,是一种X连锁疾病,由磷脂酰肌醇4,5-二磷酸5磷酸酶的活性不足引起,临床特征是先天性白内障和青光眼、严重智力低下、新生儿肌张力低下和肾脏异常。肾脏累及后出现Fanconi综合征,但终末期肾病通常要到30~40岁时出现。目前仅能对症治疗。
8.Dent病
Dent病是一种X连锁隐性遗传疾病,表现为低分子量蛋白尿、高钙尿症、肾结石、肾脏钙化和佝偻病,大多数患者的肾脏存在ClC-5氯化物通道的缺陷,干扰肾小管蛋白质重吸收,导致血尿、糖尿和氨基酸尿。
9.线粒体细胞病
是由于线粒体DNA异常导致的广泛临床异常症候群,包括神经系统疾病、色素性视网膜炎、糖尿病、胰腺功能不全、贫血、肝病和心肌病。肾脏受累可以表现为局灶节段性肾小球硬化和激素抵抗的肾病综合征,但Fanconi综合征更常见。但本病目前没有根治方法。
10.特发性Fanconi综合征
部分患者在没有任何已知病因的情况下罹患Fanconi综合征。临床表现在起病之初可不典型,而随时间推移逐步出现。大多数仍为偶发,没有家族遗传的证据。
多种物质损伤近端肾小管后可表现为不完全的Fanconi综合征、急性肾小管坏死或终末期肾病。肾小管损伤的严重程度取决于毒素的类型、摄入量和宿主的易感性。因此,对于肾小管功能障碍的患者,了解毒物暴露史和近期用药史非常重要。表1的右列为获得性Fanconi综合征的常见原因。
1.重金属
近端肾小管功能障碍的主要病因是急性重金属中毒,主要是铅和镉。铅中毒的患者肾小管功能障碍(氨基酸尿、轻度糖尿和血尿)通常被慢性肾脏病的进展以及其他器官尤其是中枢神经系统受累的表现所掩盖。与镉中毒有关的Fanconi综合征可引起严重骨痛。
2.化疗药物
许多癌症化疗药物与Fanconi综合征的肾小管功能障碍有关,尤其是顺铂和异环磷酰胺,两者的肾毒性都是剂量依赖且不可逆的。
3.其他药物和毒素
通常与GFR降低有关,包括6-巯基嘌呤、甲苯(嗅探胶)和含有马兜铃的中草药。也有丙戊酸、苏拉明、庆大霉素及雷尼替丁导致Fanconi综合征的报道。抗病毒药物,尤其是抗反转录病毒药物,如替诺福韦(tenofovir)等是导致获得性Fanconi综合征最常见的药物。
4.副蛋白血症
多发性骨髓瘤、轻链病、干燥综合征和淀粉样变性引起的副蛋白血症有时与Fanconi综合征相关,与尿液游离轻链在细胞内结晶或导致溶酶体功能障碍,进而引起近端肾小管功能障碍有关。
5.肾小球肾炎
肾病综合征很少与Fanconi综合征相关,近端小管功能障碍可发生于局灶节段性肾小球硬化(FSGS)的患者,且与预后不良相关。
(一)病因治疗
如继发者治疗基础疾病,Wilson病或重金属中毒等促进毒物排泄,遗传代谢病通过饮食管理减少代谢毒性物质沉积。
(二)对症治疗
主要针对肾脏溶质丢失及骨病。
1.酸中毒
根据碳酸氢根丢失情况补充碱剂,可用碳酸氢盐、枸椽酸盐、乳酸盐等,以血中碳酸氢根水平恢复正常为标准。
2.多尿
除针对病因如低钾血症治疗外,应补充足量的含盐液体(钾、钠、钙等)。
3.低磷血症
补充中性磷酸盐,1~3g/d使血清磷酸盐浓度正常化,如有腹泻或腹部不适,可减量。
4.骨病
许多Fanconi综合征的患者需要补充维生素D,以治疗佝偻病和骨软化症。维生素D治疗开始后,低钙血症者应补充钙。应注意补磷治疗也可加重低钙血症及骨病,故应合用维生素D。
5.肉毒碱
补充肉毒碱可改善肌肉功能和脂质代谢,但结论不一。









