


 收藏
收藏 已收藏
已收藏英文名称 :pheochromocytoma
嗜铬细胞瘤(pheochromocytoma)起源于肾上腺髓质、交感神经节或其他部位的嗜铬组织,这种瘤持续或间断地释放大量儿茶酚胺,引起持续性或阵发性高血压和多个器官功能及代谢紊乱。约10%为恶性肿瘤。本病以20~50岁最多见,男女发病率无明显差异。
嗜铬细胞瘤位于肾上腺者占80%~90%,大多为一侧性,少数为双侧性或一侧肾上腺瘤与另一侧肾上腺外瘤并存,多发性者较多见于儿童和家族性病人。肾上腺外嗜铬细胞瘤称为副神经节瘤,主要位于腹部,多在腹主动脉旁(占10%~15%),其他少见部位为肾门、肾上极、肝门区、肝及下腔静脉之间、近胰头部位、髂窝或近髂窝血管处如卵巢内、膀胱内、直肠后等。腹外者甚少见,可位于胸内(后纵隔、脊柱旁或心脏内)、颈部、颅内。肾上腺外肿瘤可为多中心的,局部复发的比例较高。
肾上腺髓质的嗜铬细胞瘤可产生去甲肾上腺素和肾上腺素,以前者为主,极少数只分泌肾上腺素,家族性者以肾上腺素为主,尤其在早期、肿瘤较小时;肾上腺外的嗜铬细胞瘤,除主动脉旁嗜铬体所致者外,只产生去甲肾上腺素,不能合成肾上腺素,因为将去甲肾上腺素转变为肾上腺素的苯乙醇胺N-甲基转移酶需要高浓度的皮质醇才能激活,只有肾上腺髓质及主动脉旁嗜铬体才具备此条件。
嗜铬细胞瘤可产生多种肽类激素,其中一部分可能引起嗜铬细胞瘤中一些不典型的症状,如面部潮红(舒血管肠肽、P物质),便秘(阿片肽、生长抑素),腹泻(血管活性肠肽、血清素、胃动素),面色苍白、血管收缩(神经肽Y)及低血压或休克(舒血管肠肽、肾上腺髓质素)等。此肿瘤还可释放嗜铬粒蛋白至血中,该蛋白血中浓度增高可协助诊断。
嗜铬细胞瘤一般呈圆形或椭圆形,有完整包膜,供应血管丰富而怒张。肿瘤体积较大,直径在一般在3~5cm,也可大于10cm,重量从小于5g至超过3500g,伴高血压的患者肿瘤平均约100g。除了发生在肾上腺髓质,腹膜后腹主动脉旁交感神经节丰富的部位也会发生,甚至见于肾、肝门、胰头、髂血管、膀胱区,以及后纵隔脊柱旁、颈部、颅内等腹腔以外部位。嗜铬细胞瘤多数为良性、单个发病,双侧或多发占少数。以往认为嗜铬细胞瘤中双侧及多发肿瘤占10%,肾上腺外肿瘤占10%,恶性肿瘤占10%。目前由于认识和诊断技术的进步,家族性嗜铬细胞瘤、多发性内分泌肿瘤相继被发现。近年统计资料表明,肾上腺内单发性嗜铬细胞瘤仅占60%~80%,双侧多发性瘤占39%~50%。
肾上腺嗜铬细胞瘤切面呈灰白或棕色,可见灶性出血、中央变性、囊性变,可伴钙化,血管丰富,间质很少,肿瘤周围有时可见正常腺体。显微镜下细胞排列呈巢状或梁状,与正常嗜铬细胞相比,肿瘤细胞较大,呈不规则多角形,细胞核多形性明显,胞质颗粒状、嗜碱性至双嗜性,因其在铬盐中颗粒着色,故此得名。嗜铬细胞瘤呈嗜铬粒素(CgA)免疫阳性,是与肾上腺皮质肿瘤和转移性非神经内分泌肿瘤鉴别的最可靠标记物。肿瘤大于6cm应高度怀疑恶性嗜铬细胞瘤,另外,肿瘤呈结节状、分叶状,切面多彩状,伴坏死、出血的斑状区域,均提示恶性可能。已经确定的恶性嗜铬细胞瘤组织学标准包括:①包膜侵犯:②侵犯血管;③扩散到肾上腺周围脂肪结缔组织:④膨胀的、大的、融合性细胞巢;⑤弥漫性生长、坏死;⑥细胞成分增加;⑦肿瘤细胞呈梭形;⑧细胞核重度多形性,瘤细胞单一性(小细胞、核/质比率高);⑨核深染、大核仁,核分裂增多;⑩任何非典型核分裂象。没有一个组织学特征能独立确定嗜铬细胞瘤的恶性倾向。实际上,最严格的恶性定义是转移必须出现在原来没有嗜铬组织的部位。
下面介绍几种特殊类型的嗜铬细胞瘤。
1.多发性内分泌瘤(multiple endocrine neoplasia,MEN)
是一种常染色体显性遗传病。临床表现为多种内分泌病症的组合,即多发性内分泌腺瘤综合征(MENS)。1964年,Pear将广泛存在于内分泌腺体及其他组织中能产生内分泌多肽物质的细胞统称为APUD细胞,这些细胞来源于神经嵴,分布于垂体、甲状腺、甲状旁腺、胰腺、肾上腺髓质。当神经嵴细胞发育异常时,就会发生MEN,有的学者将这种肿瘤统称为APUD瘤。根据各种内分泌腺瘤发病的不同,MEN分为3型:①MENⅠ型:又称Wermer综合征,包括垂体、甲状旁腺和胰腺的肿瘤。②MENⅡ型:与RET密码子634突变相关。MENⅡa型:又称Sipple综合征,包括嗜铬细胞瘤或肾上腺髓质增生并甲状腺髓样癌、甲状旁腺肿瘤。MENⅡb型:除MENⅡa型肿瘤外,还可发生多发性皮肤或黏膜神经瘤、马方样体型等。③MENⅢ型:甲状旁腺瘤和乳头状甲状腺癌。也有人把MENⅡb型与MENⅢ型合在一起。
2.Von Hippel-Lindau(VHL)病
是由VHL肿瘤抑制基因种系突变引起的显性遗传性家族性癌综合征,又称VHL综合征。该病有明显的表型变异性和与年龄相关的外显率,常见的肿瘤包括视网膜和中枢神经系统血管网状细胞瘤、肾细胞癌、嗜铬细胞瘤和胰腺内分泌肿瘤,其中嗜铬细胞瘤的总发病率为10%~30%。VHL病根据临床表现再分成亚型,其分类如表1。
表1 VHL病临床分类
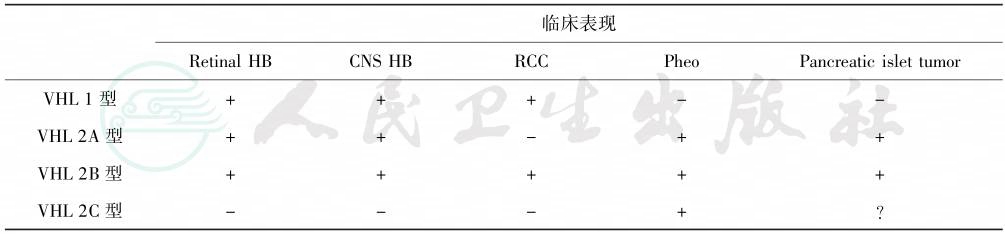
注:Retinal HB:视网膜血管网状细胞瘤;CNS HB:中枢神经系统血管网状细胞瘤;RCC:肾细胞癌;Pheo:嗜铬细胞瘤;Pancreatic islet tumor:胰岛细胞瘤。
引自:实用外科学(全2册).第4版.ISBN:978-7-117-23988-2.主编:
3.神经纤维瘤病1型(neurofibromatosis type1, NF 1)
又称Von Recklinghausen病,NF1是常染色体显性遗传性疾病,以皮肤神经纤维瘤、皮肤色素沉着(牛奶咖啡斑)和骨发育不良为特征。约1%的患者可发生嗜铬细胞瘤,好发于40~50岁年龄段。值得注意的是,约5%的嗜铬细胞瘤患者合并有NF1,这类患者的遗传学类型更常伴有MEN 2或VHL病。
以上3种疾病均称为家族性嗜铬细胞瘤(familialpheochromocytoma),与线粒体复合体Ⅱ基因SDHD、SDHB、SDHC突变有关,占嗜铬细胞瘤的6%~10%。家族性嗜铬细胞瘤具有以下特点:①是常染色体显性遗传疾病,有高度外显率;②发病年龄较早,可见于儿童;③47%多为双侧发病,多为两个以上的内分泌腺体受累。双侧性嗜铬细胞瘤中约50%为家族性;④同一家族中的发病的患者,其发病年龄和肿瘤部位往往相同;⑤常与MENⅡ型或神经外胚层发育异常,如神经纤维瘤病(NF)、视网膜血管瘤(Von Hippel)、脑脊髓血管网状细胞瘤(Lindiau)等相伴发,国内外均有相继报道;⑥有较高的复发率。
4.多种内分泌功能性嗜铬细胞瘤
即嗜铬细胞瘤具有分泌两种以上的内分泌激素的功能。1985年Shanberg证实嗜铬细胞可自主性分泌异位性甲状旁腺素,并发高钙血症。1979年Forman及Spark等首先报道嗜铬细胞瘤异位分泌促皮质激素(ACTH),与肺癌及其他肿瘤所分泌的大形ACTH不同,70%为小形ACTH,是人类标准的ACTH,可引起典型的库欣综合征,如术前未能确诊,术后未加重视,手术死亡率达50%以上。嗜铬细胞瘤还可分泌α-MSH、血管活性肠肽(vasoactive intestinal peptide)、前列腺素以及神经系统所具有的P-物质、γ神经肽、生长抑素(somatostatin)等物质,其临床意义有待进一步确定。
5.特殊部位的嗜铬细胞瘤
(1)肾门部的嗜铬细胞瘤:
多见于左侧。肿瘤直接浸润压迫可引起肾动脉狭窄;高儿茶酚胺血症直接引起肾动脉痉挛性收缩,随着病程延长,动脉壁发生纤维性变及增生,从而发生肾动脉解剖性狭窄。因此,这类病例的肾素系统也呈活跃状态。在高儿茶酚胺和高肾素的双重作用下,恶性高血压的进展非常迅猛。
(2)胰腺后方的嗜铬细胞瘤:
常位于腹主动脉及下腔静脉之间,易向血管内浸润,界限不清,处理极为困难。
(3)膀胱嗜铬细胞瘤:
是副神经节瘤的一种。症状常常在排尿时发作是其重要特点。肿瘤位于膀胱肌壁间,多不侵犯黏膜。
嗜铬细胞瘤手术切除前采用α受体拮抗药使血压下降,减轻心脏的负担,并使原来缩减的血管容量扩大。常用的α受体拮抗药为作用较长(半衰期36小时)的酚苄明,开始时每日口服2次,每次10mg,按需逐渐加量至血压得到控制。不良反应为直立性低血压,鼻黏膜充血。选择性的α受体拮抗药哌唑嗪、多沙唑嗪也可获满意效果,并可避免全部α受体拮抗的不良后果,如明显的低血压和心动过速。半衰期较短,可较灵活调节用量。起始用小剂量以避免严重的直立性低血压。哌唑嗪起始口服0.5mg或1mg,了解病人对此药的敏感性,以后按需增加,剂量每次2~4mg,日服2~3次。多沙唑嗪每日用量2~8mg,控释剂每片4mg,每日1次,1~2片,必要时可加量。
当病人骤发高血压危象时,应积极抢救:立即静脉缓慢推注酚妥拉明(phentolamine,regitine)1~5mg,同时密切观察血压,当血压下降至160/100mmHg左右即停止推注,继之以10~15mg溶于5%葡萄糖生理盐水500ml中缓慢静脉滴注。也可舌下含服钙通道阻滞药硝苯地平10mg,以降低血压。
在手术治疗前,α受体拮抗药的应用一般不得少于2周,并进正常或含盐较多的饮食(心衰者除外),以使原来缩减的血容量恢复正常。虽然酚苄明作用时间较长,仍宜用到手术前一日为止,以免手术时出现血压骤升。术前β受体拮抗药不必常规应用,如病人有心动过速或心律失常则需采用。在用β受体拮抗药之前,必须先用α受体拮抗药使血压下降,如单独用β受体拮抗药,则由于阻断β受体介导的舒血管效应而使血压升高,甚而发生肺水肿,尤其是分泌肾上腺素为主的病人。
切除嗜铬细胞瘤有一定危险性,必须在富有经验的外科医师和麻醉师主持下施行。在麻醉诱导期,手术过程中,尤其在接触肿瘤时,可出现血压急骤升高和(或)心律失常。对血压骤增者,可采用速效的α受体拮抗药酚妥拉明静脉推注,继之以静滴或用硝普钠静滴。对心律失常者,可用β2受体拮抗药或其他抗心律失常药,如利多卡因等。肿瘤被切除后,血压一般降至90/60mmHg。如血压低,周围循环不良,表示血容量不足,应补充适量全血或血浆,必要时也可静脉滴注适量去甲肾上腺素,但不可用缩血管药来代替补充血容量。
嗜铬细胞瘤切除后,血压多能恢复正常,但在手术后第1周,血压仍可偏高,同时血、尿儿茶酚胺也可偏高。因此,在手术后1个月左右,应根据血压状态和血、尿儿茶酚胺,方能更准确地判断治疗效果。小部分病人手术后仍有高血压,可能因合并原发性高血压,或儿茶酚胺长期增多损伤血管所致。由于嗜铬细胞瘤有可能为多发性或复发性,故术后应随访观察。
恶性嗜铬细胞瘤的治疗较困难,一般对放疗和化疗不敏感,可用抗肾上腺素药对症治疗。链佐星治疗的效果不一,也可用酪氨酸羟化酶抑制剂α-甲基间酪氨酸阻碍儿茶酚胺的生物合成,131I-MIBG治疗可获一定效果。已发生转移的恶性嗜铬细胞瘤预后不一,重者在数个月内死亡,少数可活10年以上,5年生存率约为45%。转移最常见的部位为骨骼、肝、淋巴结、肺,其次为脑、胸膜、肾等。









