


 收藏
收藏 已收藏
已收藏血液苯丙氨酸浓度持续高于2mg/dl(120μmol/L)称为高苯丙氨酸血症(hyperphenylalaninemia)。遗传性高苯丙氨酸血症患儿血液苯丙氨酸持续性高浓度,高于6mg/dl(360μmol/L)。
遗传性高苯丙氨酸血症包括两类遗传缺陷。一类为苯丙氨酸羟化酶(phenylalanine hydroxylase,PAH)缺陷所致经典型苯丙酮尿症(phenylketonuria,PKU)和高苯丙氨酸血症,占90%以上;另一类为PAH的辅酶四氢生物蝶呤(tetrahydrobiopterin,BH4)的代谢缺陷所致四氢生物蝶呤缺乏症。两类缺陷均导致苯丙氨酸代谢障碍,体内苯丙氨酸异常蓄积,引起一系列神经系统损害。但两类疾病的诊断与治疗方法不同,应及早鉴别(表1)。
表1 遗传性高苯丙氨酸血症的分类、鉴别与治疗
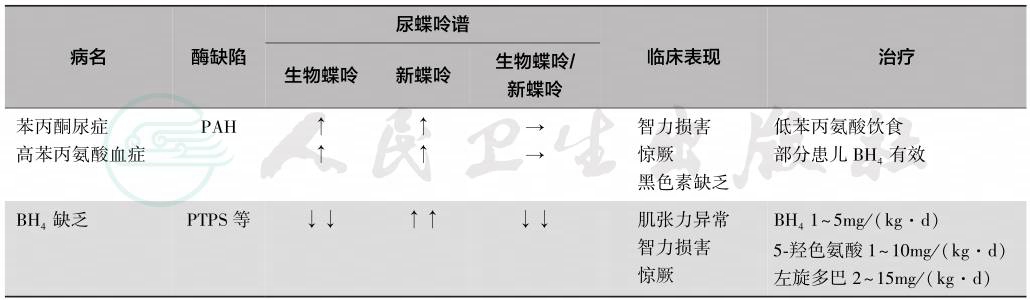
注:BH4:四氢生物蝶呤。
PKU患儿肝PAH的水平仅有正常人的1%或更低,经食物摄取的蛋白质降解后产生的苯丙氨酸不能代谢转化,体内苯丙氨酸、苯丙酮酸、苯乙酸蓄积,酪氨酸、黑色素、肾上腺素等生物活性物质缺乏,引起神经髓鞘发育障碍、神经精神异常。四氢生物蝶呤缺乏症患儿缺乏四氢生物蝶呤等神经递质,导致肌张力异常、癫痫发作、免疫力下降。
PKU是常染色体隐性遗传代谢病,由于PAH基因突变导致苯丙氨酸代谢障碍。PAH位于12q23.2,含13个外显子,国内外已报告了近千种基因突变,具有高度遗传异质性,突变类型与人种、民族、临床特点有一定的关系。我国PKU患病率较高,一般人群中PAH基因杂合突变携带者高达1/50~1/30。
(一)临床表现
PKU的主要危害为神经精神损害。未经治疗的患儿在新生儿期多无明显症状,数月龄时出现轻重不同的智力发育落后,近半数患儿合并癫痫,其中婴儿痉挛症占1/3。大多数患儿有烦躁、易激惹、抑郁、多动、孤独症倾向等精神行为异常,最终将造成中度至极重度智力障碍。由于黑色素缺乏,患儿毛发逐渐变黄,皮肤白,虹膜颜色浅。旁路代谢产物苯丙酮酸、苯乙酸自尿液、汗液中大量排出,常有鼠尿样体臭。
必须重视的是,PKU患儿在新生儿期和婴儿早期多无明显异常,部分患儿有呕吐、喂养困难、烦躁等非特异性症状,临床表现个体差异较大,极易漏诊或误诊,只有通过新生儿筛查才能早期发现。
(二)辅助检查
1.新生儿筛查或高危筛查,血苯丙氨酸显著增高,>120μmol/L(2mg/dl)及苯丙氨酸/酪氨酸比值>2.0。苯丙氨酸>360μmol/L(6mg/dl),经低苯丙氨酸饮食控制后下降。
2.尿蝶呤谱正常,可鉴别四氢生物蝶呤缺乏症。
3.红细胞二氢蝶啶还原酶活性正常,可鉴别二氢蝶啶还原酶缺乏症。
4.基因诊断 PAH双等位基因突变。
5.四氢生物蝶呤负荷试验 约30%的PKU患儿服用四氢生物蝶呤后血液苯丙氨酸浓度下降。
6.脑影像学检查 一些疾病控制不良的患儿可见脑白质异常。
(三)诊断与鉴别诊断
1.对新生儿筛查或临床高危筛查血苯丙氨酸增高者,建议采用定量法(荧光法或串联质谱法)测定,苯丙氨酸浓度>120μmol/L及苯丙氨酸/酪氨酸比值>2.0确诊为高苯丙氨酸血症。
2.临床患儿出现智力发育落后、皮肤和毛发色浅淡,汗液和尿液有鼠臭味,血苯丙氨酸浓度>120μmol/L及苯丙氨酸/酪氨酸比值>2.0者可确诊。
3.鉴别肝病及其他疾病导致的继发性高苯丙氨酸血症,任何疾病导致的先天或后天肝损害患儿血液苯丙氨酸均可轻度增高,需要通过病因调查、血液氨基酸分析等生化分析、基因分析鉴别诊断。
(四)治疗与预后
1.治疗时期
PKU一旦确诊,应立即开始饮食或药物干预,终身治疗。开始治疗的年龄越小,预后越好。如能在症状开始前治疗,绝大多数PKU患儿可以获得正常发育,与同龄人一样就学就业、结婚生育。新生儿筛查是早期发现PKU的重要措施,2019年我国新生儿PKU筛查覆盖率达到了98%。如果在发病后开始治疗,多数患儿将遗留不可逆性脑损害。
2.低苯丙氨酸饮食
是治疗PKU的主要方法,限制天然蛋白质摄入,以防止苯丙氨酸及其代谢产物的异常蓄积,补充无或低苯丙氨酸配方奶粉,满足机体蛋白质、热量等营养需要,保证患儿的正常发育。血中苯丙氨酸浓度应控制在理想范围(2~6mg/dl,120~360μmol/L),苯丙氨酸浓度过高或过低都将影响生长发育。待血苯丙氨酸降至理想浓度时,可逐渐少量添加天然饮食,首选母乳。较大婴儿及儿童可添加低蛋白低苯丙氨酸食物及少量牛奶、粥、面、蛋等。
3.四氢生物蝶呤
近30%的PKU患儿为四氢生物蝶呤反应型,经四氢生物蝶呤[1~20mg/(kg·d)]治疗后血液苯丙氨酸浓度显著降低。部分患儿只需口服四氢生物蝶呤即可获得良好的控制,部分患儿在服用四氢生物蝶呤的基础上,对天然蛋白质的耐受性提高,可以减少低苯丙氨酸配方奶粉食用量[3,4]。
4.肝移植
对于饮食控制困难的PKU患儿,肝移植可以实现根治。
(五)预防
新生儿筛查是早期发现高苯丙氨酸血症的重要措施,如果在发病后开始治疗,患儿可能遗留不可逆性脑损害。如能在症状前开始治疗,绝大多数患儿可以获得正常发育,与同龄人一样就学就业、结婚生育。
对于基因诊断明确的家系,可在母亲下一次妊娠8~13周左右留取胎盘绒毛,或在妊娠16~22周抽取羊水,分取羊水细胞,通过胎儿PAH基因分析进行产前诊断。
四氢生物蝶呤缺乏症(tetrahydrobiopterin deficiency)又称异型PKU,约占遗传性高苯丙氨酸血症的5%~10%,中国南方多于北方。
(一)病因与发病机制
已发现六种酶缺陷与四氢生物蝶呤生成障碍有关,其中6-丙酮酰四氢蝶呤合成酶(6-pyruvoyl tetrohydropterin synthase,PTPS)缺乏症最常见,二氢蝶啶还原酶(dihydropteridine reductase,DHPR)缺乏症次之,其余较为少见。编码6-丙酮酰四氢蝶呤合成酶的PTS基因位于11q23.1,含6个外显子,编码DHPR的基因QDPR位于4p15.32,含有7个外显子,均已发现多种致病突变。
四氢生物蝶呤是PAH、酪氨酸羟化酶和色氨酸羟化酶的辅酶,不仅参与苯丙氨酸的代谢,也参与多巴、肾上腺素、5-羟色氨酸的合成,具有多种生物作用。四氢生物蝶呤缺乏症导致高苯丙氨酸血症,同时引起多巴、肾上腺素、5-羟色氨酸等生理活性物质缺乏,神经细胞髓鞘蛋白合成下降,机体免疫功能下降。
(二)临床表现
四氢生物蝶呤缺乏症患儿出生时正常,无特异性症状与体征,临床诊断困难。与PAH缺乏症导致的高苯丙氨酸血症患儿相比,患儿多自婴儿期出现惊厥、发育落后、吞咽困难、肌张力异常、松软或角弓反张。低苯丙氨酸饮食治疗无效,即使食用特殊奶粉后血苯丙氨酸浓度降至正常,神经系统损害仍进行性加重。四氢生物蝶呤参与免疫机制,患儿抵抗力较差,易感染,多死于肺炎等感染性疾病。
(三)辅助检查
1.血苯丙氨酸增高
新生儿筛查或高危筛查,血液苯丙氨酸可波动在2~20mg/dl(120~1 200μmol/L),经治疗后下降。
2.尿蝶呤谱异常
各型酶缺乏患儿尿蝶呤谱有所不同,PTPS缺乏症患儿尿新蝶呤浓度明显增高,生物蝶呤浓度降低,新蝶呤/生物蝶呤显著增高;DHPR缺乏症患儿尿新蝶呤、生物蝶呤均增高,新蝶呤/生物蝶呤正常;GTPCH1缺乏症患儿尿新蝶呤、生物蝶呤浓度均降低,两者比例正常,有助于鉴别。
3.红细胞二氢蝶啶还原酶(DHPR)活性测定
DHPR缺乏症患儿酶活性低下。
4.四氢生物蝶呤负荷试验
对于血液苯丙氨酸基础浓度>6mg/dl(360μmol/L)的患儿,给予四氢生物蝶呤20mg/kg,负荷前、负荷后1小时、2小时、4小时、8小时取血测定血苯丙氨酸浓度,负荷前、负荷后4~8小时留尿进行蝶呤谱分析。四氢生物蝶呤缺乏症患儿常于负荷后4~8小时血苯丙氨酸浓度降至正常,而PAH缺乏所致经典型PKU和高苯丙氨酸血症患儿血苯丙氨酸浓度无明显下降。
5.基因诊断
确定患儿的致病基因突变,明确基因诊断,如PTPS、DHPR等基因缺陷。
(四)诊断与鉴别诊断
对所有新生儿筛查或临床高危筛查中血苯丙氨酸增高的患儿,通过尿蝶呤谱分析、红细胞DHPR活性测定和基因分析进行确诊及分型。
(五)治疗与预后
一旦确诊,应立即开始治疗,以预防或减轻神经系统损害,终身治疗。
1.四氢生物蝶呤
各型四氢生物蝶呤缺乏症方法不同,PTPS缺乏症患儿四氢生物蝶呤剂量为1~5mg/(kg·d),根据体重、血苯丙氨酸浓度及尿蝶呤谱分析等调节剂量。
2.神经递质前质补充治疗
如左旋多巴、5-羟色氨酸。
3.低苯丙氨酸饮食治疗
对于DHPR缺乏症患儿,需要限制天然蛋白质,补充特殊奶粉,并补充亚叶酸,以防治脑叶酸缺乏症[5-7]。
(六)预防
新生儿筛查是早期发现四氢生物蝶呤缺乏症的重要措施,如果在发病后开始治疗,患儿可能遗留不可逆性脑损害。如能在症状前开始治疗,绝大多数四氢生物蝶呤缺乏症患儿可以获得正常发育,与同龄人一样就学就业、结婚生育。
对于基因诊断明确的家系,可在母亲下一次妊娠8~13周左右留取胎盘绒毛,或在妊娠16~22周抽取羊水,分取羊水细胞,通过胎儿基因分析进行产前诊断。









