


 收藏
收藏 已收藏
已收藏英文名称 :hereditary coproporphyria
遗传性粪卟啉病(hereditary coproporphyria,HCP)以常染色体显性遗传方式导致粪卟啉原氧化酶(coproporphyriongen oxidase,COPRO’gen oxidase,CPOX)缺陷而发病。2/3患者可无症状。急性发作常由药物诱发。神经症状与急性间歇性卟啉病相似,但相对较轻。皮肤光敏症状比混合性卟啉病少见。与先天性红细胞生成性卟啉病主要含粪卟啉Ⅰ型异构体不同,本病排泄的粪卟啉约有95%是粪卟啉Ⅲ。急性发作时ALA和卟胆原的排泄也可增多。
关于卟啉病的流行病学,目前尚缺乏大规模的临床资料。一般而言,临床常见的卟啉病类型为急性间歇性卟啉病(acute intermittent porphyria,AIP)、迟发性皮肤卟啉病(porphyria cutanea tarda,PCT)、HCP和混合性卟啉病(miscellaneous porphyria,MP),以AIP发病率最高,约为1/10万;低于10%的急性卟啉症患者会出现急性发作,约90%终生不发病。冯加纯等统计国内累计报道的卟啉病732例,结果AIP占94.44%,MP占5.56%。发病年龄3~68岁,平均30.61岁±11.77岁;男女比例为1∶1.72;有遗传史的占6.42%。
遗传性粪卟啉病任何年龄均可发病,多数在青春期后发病,成年女性患者居多,有明显家族史。多数患者无症状,少数呈间隙性发作。约30%患者有光敏性皮损,少数患者仅尿、粪中粪卟啉排出增多。本型可因巴比妥类药物、激素类药物及解痉药而诱发,饥饿、妊娠可使病情加重。
血卟啉病的发病与遗传因素有关,多数为常染色体显性遗传,仅少数类型为隐性遗传。某些药物,如磺胺、巴比妥类、氯化奎宁、麻醉药等,以及中毒、感染、饮酒、劳累、精神刺激、月经和妊娠、某些疾病如肝肿瘤等都可诱使本病发作。
卟啉代谢及血红素生物合成的速度与ALA合成酶的活性相互形成负反馈调节。当血红素生物合成障碍时负反馈调节作用丧失,ALA合成酶作用失去调控,致使卟啉中间产物产生过多。每一型卟啉病均与血红素合成途径中的一种酶缺陷有关。1986年首次证实与卟啉病相关的酶缺陷系由相应的基因突变所致;通过对酶的编码基因和cDNA进行克隆,可以确定导致酶缺陷的相应基因突变。
根据代谢紊乱出现的部位,卟啉病可分为肝原性和红细胞生成性血卟啉病;根据临床表现和病程经过,分为急性和慢性皮肤型。根据特异酶的缺陷,可分为ALA脱水酶缺乏性卟啉病(ALA anhydrase deficiency porphyria,AADP)、急性间歇性卟啉病、先天性红细胞生成性卟啉病(congenital erythropoietic porphyria,CEP)、迟发性皮肤型卟啉病、肝红细胞生成性卟啉病、遗传性粪卟啉病、混合性卟啉病和原卟啉病(protoporphyria,PP)八种类型(图1)。
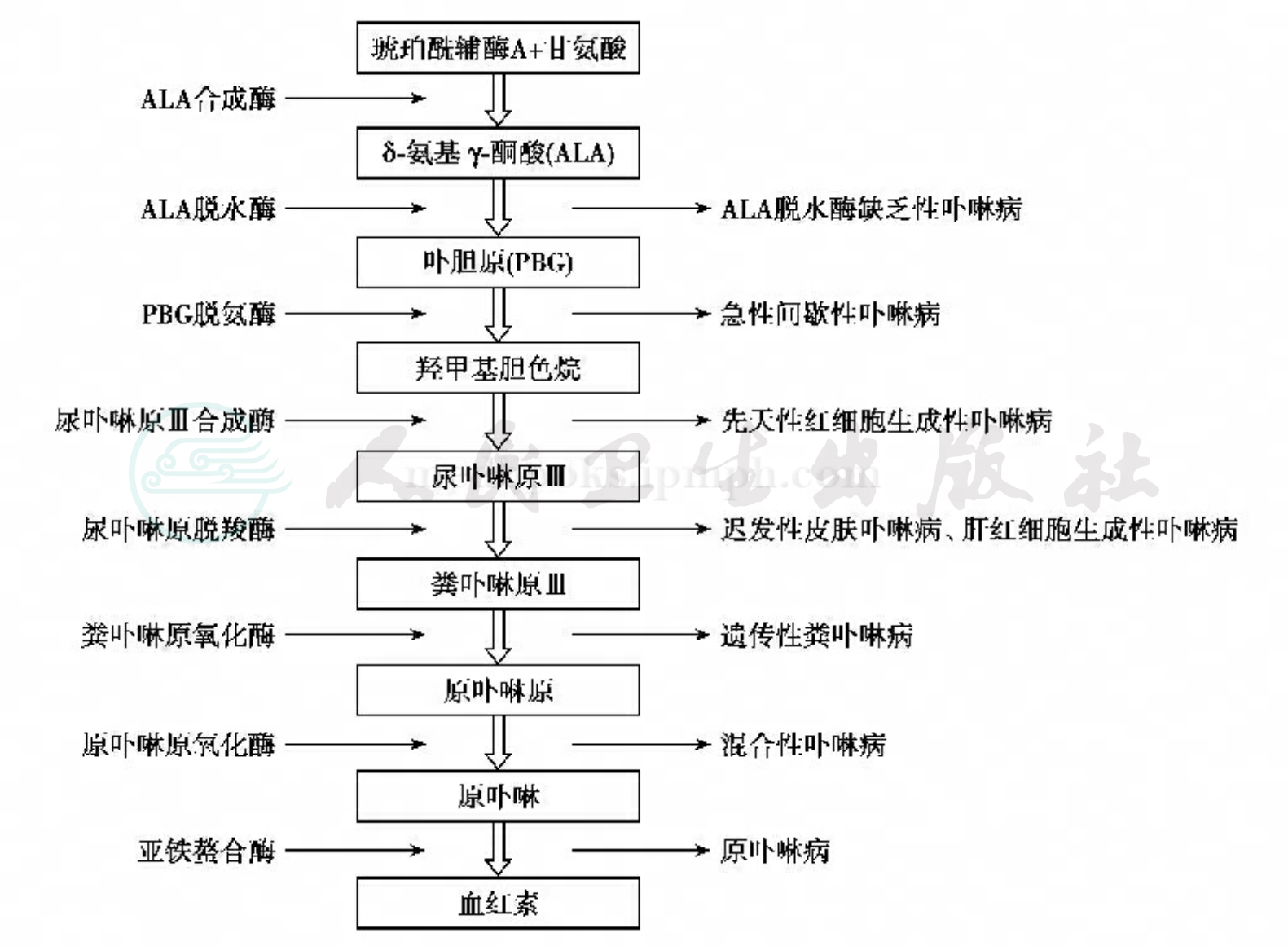
图1血红素生物合成途径及其相应酶缺乏导致的各型卟啉病
卟啉是以四吡咯环螯合铁为基本结构的色素总称,是组成血红蛋白、肌红蛋白、过氧化物酶、细胞色素以及触酶的辅基成分。其基本化学结构是由四个吡咯环的四个甲烯桥联合成为一个总环,每个吡咯环上的氢原子为不同化学基团所取代时便成为各种不同的卟啉,其中与人类血红素合成有关的包括尿卟啉、粪卟啉以及原卟啉(图2)。卟啉及前体物质的分泌主要取决于化合物的溶解性,水溶性化合物分泌入尿液,而非水溶性的则分泌入胆汁。卟啉前体δ‐氨基γ‐酮酸、卟胆原及尿卟啉是水溶性,主要经尿液排泄;原卟啉难溶于水,几乎全部分泌入胆汁;粪卟啉既可分泌入胆汁也可分泌入尿液。
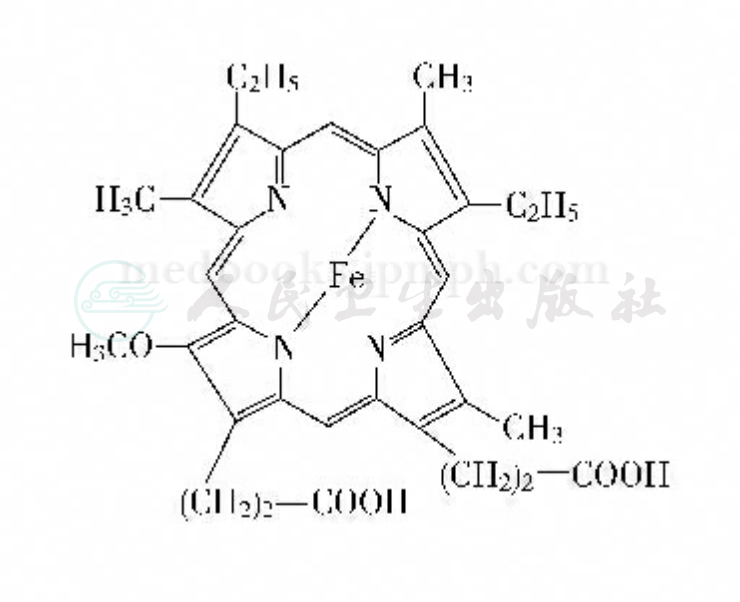
图2卟啉环分子结构
实验室检查的特点是患者的尿液和粪便中含有大量的粪卟啉。
治疗措施包括去除诱发因素,输注高铁血红素和补充葡萄糖等。
本病应预防为主。避免近亲结婚,戒烟戒酒,女性患者不宜妊娠分娩。避免过劳、感染、精神刺激等诱因,忌用巴比妥、酒精、雌激素、磺胺以及氯霉素等,对于有皮损的患者应避免日晒,应用遮光剂。由于本病为先天性遗传病,一旦诊断某家族成员患病,建议所有亲属行相关检查以明确是否为基因携带或为隐匿型。









