


 收藏
收藏 已收藏
已收藏英文名称 :copper deficiency
铜是人和动物所必需的营养素之一,是许多神经代谢酶(如氧化酶、羟化酶、超氧化物歧化酶等)的组成部分,许多疾病都可导致铜缺乏并出现相应的临床表现。体内最丰富的含铜酶为超氧铜化物歧化酶,可以发挥自由基清除的作用从而防止细胞膜氧化损伤,其次含铜酶为血浆铜蓝蛋白,是血浆中铜的主要转运体,也是铁从肝中释放和与转铁蛋白结合的必需元素。
足月新生儿体内铜总量约20mg,约1/2是在妊娠后3个月从母体获得的,其中50~60%贮存于肝(成人仅占5%),可供生后6个月的需要。婴幼儿每日铜需要量约80μg/kg,年长儿40μg/kg,成人2~5mg。含铜丰富的食物为肝、肾、甲壳类、谷类、黄豆、硬壳果等。正常饮食的成人及儿童不易缺铜。但乳类含铜较少,人乳为 400μg/L(初乳 600μg/L),牛乳300μg/L,单纯以乳类喂养的婴儿,尤其是早产儿(每日最低铜需要量约100μg/kg),易于发生铜缺乏症(copper deficiency)。
1.营养性铜缺乏症
(1)先天贮存不足:
多见于早产儿,特别是VLBW儿,由于铜储存不足和生后进食的奶量较少,而且人乳和一般配方奶的铜含量较低,使铜的摄入量很少。
(2)摄入不足:
长期单纯乳类喂养;长期全胃肠外静脉营养,未补充铜剂。
(3)吸收障碍:
长期腹泻、吸收不良综合征、小肠大段切除(短肠综合征)。
(4)其他:
严重营养不良或肾病综合征时肝产生铜蓝蛋白减少或经肾排出增多。
2.遗传代谢病
Menkes综合征,为性联隐性遗传。
体内的铜参与30余种酶的构成,在许多代谢途径中具有重要作用,缺铜时可发生广泛的代谢、功能障碍和病理改变。
1.血浆铜蓝蛋白
①血浆铜的90%~95%与铜蓝蛋白结合,运送给红细胞、骨髓和需铜组织。铜缺乏时血浆铜蓝蛋白减少,使铜供应不足;②铜蓝蛋白具有亚铁氧化酶的作用,可将从肠吸收和从贮存部位释放的Fe2+氧化为Fe3+,然后铁才能与运铁蛋白结合而转运。铜缺乏时铁的吸收和转运障碍,可产生小细胞低色素性贫血。③具有多酚氧化酶的作用,可氧化某些二胺及多酚如去甲肾上腺素、肾上腺素和5-羟色胺等,对调节神经递质和神经系统的功能有重要影响。
2.δ-氨基-γ-酮戊酸(ALA)脱水酶
是将ALA催化为胆色素原所需的酶,再经过其他酶的连续催化,生成原卟啉;在亚铁螯合酶作用下,与Fe2+结合成血红素;最后与珠蛋白构成血红蛋白。缺铜时原卟啉、血红素及血红蛋白生成减少。而血红素又是构成肌红蛋白、细胞色素、过氧化物酶、过氧化氢酶和尿黑酸氧化酶的必要成分。
3.细胞色素氧化酶
能源物质在体内氧化过程中,通过细胞内线粒体的电子传递系统(呼吸链),在多种酶和辅酶的连锁反应下,将代谢物脱下的氢逐步传递,产生能量,最后与氧化合成水。所产生大量能量的40%~50%以上用于将ADP磷酸化生成ATP贮存,小部分用于线粒体内离子转移和代谢更新,其余以热的形式发散。细胞色素氧化酶是呼吸链最后阶段所需的酶,缺乏时将发生能量代谢障碍,影响细胞功能,造成神经系统等的损害。
4.超氧化物歧化酶
在氧化应激反应时,形成超氧阴离子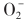 ,可使组织损伤。正常情况下被超氧化物歧化酶催化,
,可使组织损伤。正常情况下被超氧化物歧化酶催化,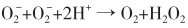 ,生成的H2O2进一步分解。缺铜时可发生神经脱髓鞘,认为与此酶缺乏有关。
,生成的H2O2进一步分解。缺铜时可发生神经脱髓鞘,认为与此酶缺乏有关。
5.多巴胺-β-羟化酶
可将多巴胺催化为去甲肾上腺素,进而转化为肾上腺素。是缺铜时发生神经系功能障碍的原因之一。
6.赖氨酰氧化酶
是胶原和弹性蛋白分子内和分子间形成共价键而紧密交联所需的酶。缺乏时共价键形成障碍,血管壁的胶原纤维与弹性纤维的张力和弹性减低甚至断裂,可导致血管扩张或破裂。骨有机基质损害,可发生类似婴儿维生素C缺乏的骨骼病变。
7.酪氨酸酶
可将二羟苯丙氨酸(DOPA)转化为黑色素。缺乏时皮肤及头发黑色素形成障碍。
1.血常规检查
红细胞,血红蛋白,网织红细胞,白细胞及中性粒细胞均降低,血小板正常。多为小细胞低色素性贫血。
2.骨髓检查
红细胞和粒细胞系统增生减低,成熟障碍,红细胞及粒细胞细胞质中有空泡形成,铁粒幼红细胞增加。
3.生化检查
(1)血清铜:
正常成人为10.9~21.08μmol/L(69.4~134.3μg/dl),足月儿为 3.14~10.99μmol/L(20~70μg/dl)。早产儿更低,胎龄 35~36 周为(6.15±2.17)μmol/L[39.2±13.8(μg/dl)],25~28 周仅为(4.49±2.68)μmol/L[28.6±17.1(μg/dl)]。足月儿于生后1个月增至成人水平,早产儿迟至4个月才达到此水平,可能与肝产生铜蓝蛋白的功能不成熟有关。成人低于 10.9μmol/L(70μg/dl)、新生儿低于 5.4μmol/L(35μg/dl)提示缺铜[19]。
(2)血浆铜蓝蛋白:
正常成人为 0.93~2.65μmol/L(14~40mg/dl),新生儿为 0.06~1.99μmol/L(1~30mg/dl);生后与血清铜并行增高达成人水平。当低于1μmol/L(15mg/dl)通常见于铜降低。
此外,有学者认为检测血清IL-2水平等可作为了解多种动物体内铜状态的标志物,但其在人体内的意义有待于进一步研究。
除治疗原发病外,给予0.5%硫酸铜0.2~0.6ml(1~3mg),分次口服。Menkes综合征患儿由于小肠吸收的铜转运障碍,需胃肠道外给药,一般将硫酸铜1~2mg溶于50~100ml生理盐水,静脉滴注,每3~4天1次,需在生后早期治疗,否则神经系统等的受累难以恢复。为预防缺铜,建议给小早产儿补充元素铜数月,按100~500μg/d,对完全静脉营养的患儿,在营养液中加元素铜 20~30μg/(kg·d)。硫酸铜中元素铜含量为39.82%,即1mg含有400μg元素铜。









