


 收藏
收藏 已收藏
已收藏英文名称 :congenital biliary dilatation
中文别名 :胆管扩张症
先天性胆管扩张症(congenital biliary dilatation)可发生于肝内、肝外胆管的任何部分,因好发于胆总管,曾称之为先天性胆总管囊状扩张,现在认为应称为胆管扩张症。本病女性多于男性,男女比约为1∶(3~4),约80%病例在儿童期发病。
胆管壁先天性发育不良及胆管末端狭窄或闭锁是发病的基本因素,可能原因有:①先天性胰胆管合流异常:胚胎期胆总管和胰管是分开的,如果胆总管以直角进入胰管,或胰管在壶腹上方汇入胆管,胰液反流入胆管致内膜受损并发生纤维性变,导致胆总管囊性扩张;②先天性胆道发育不良:胚胎期,原始胆管增殖为索状,以后再空泡化贯通,如胆管上皮过度空泡化,可致胆管壁薄弱而发生囊性扩张;③遗传因素:本病女性发病率明显高于男性,有人认为与性染色体有关。
根据胆管扩张的部位、范围和形态,分为五种类型(图1)。
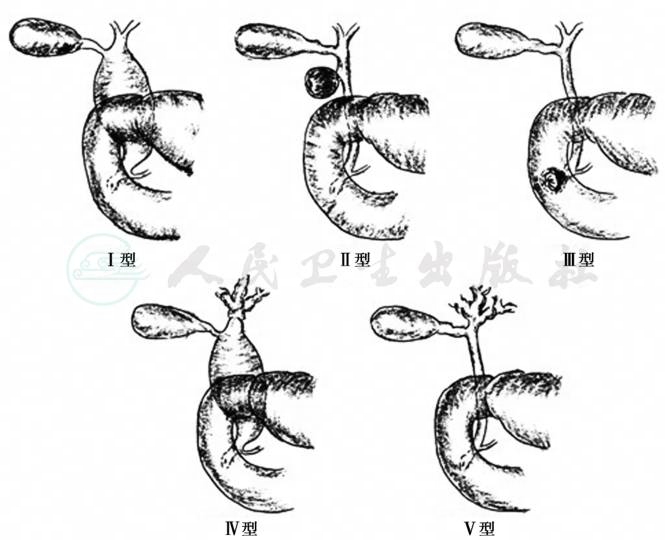
图1 先天性胆管扩张症的分型
Ⅰ型:囊性扩张。最常见,约占90%。可累及肝总管、胆总管的全部或部分,胆管呈球状或葫芦状扩张,直径最大者25cm,扩张部远端胆管严重狭窄。胆囊管一般与囊状扩张汇合,其左右肝管及肝内胆管正常。
Ⅱ型:憩室样扩张。为胆总管侧壁局限性扩张呈憩室样膨出,少见。
Ⅲ型:胆总管十二指肠开口部囊性突出。胆总管末端十二指肠开口附近囊性扩张,囊状扩张进入十二指肠腔内致胆管部分梗阻。
Ⅳ型:肝内外胆管扩张。肝内胆管有大小不一的多发性囊性扩张,肝外胆管亦呈囊性扩张。
Ⅴ型:肝内胆管扩张(Caroli病)。肝内胆管多发性囊性扩张伴肝纤维化,肝外胆管无扩张。
扩张囊壁常因炎症、胆汁潴留而引起溃疡,甚至癌变,其癌变率为10%,成人接近20%,较正常人群高出10~20倍。囊性扩张的胆管腔内也可有胆石形成,成年人中合并胆石者可高达50%。
胆管扩张可发生于肝内、肝外的任何部位,基本上是囊状扩张和梭状扩张两种形态。常见型是胆总管囊状扩张,肝内胆管不扩张或有多发囊状扩张,而扩张以下胆管显著狭小,仅有1~2mm直径,胆管狭窄部位在胰外的游离胆总管与胰内胆总管的移行部,由于梗阻而致近侧胆管内压增高而导致囊形扩张和管壁增厚,合流形态为胆管→胰管合流型。胆总管梭状扩张病例的肝内胆管扩张至末梢胆管渐细,其狭窄部位在两管合流部和胰胆共通管的十二指肠壁内移行部两处,由于梗阻而致共通管轻度扩张和胆总管梭状扩张,合流形态为胰管→胆管合流型。发病时胆管扩张明显,症状缓解时略见缩小。
按病程的长短,扩张管壁可呈不同的组织病理变化,在早期病例,管壁呈现反应性上皮增生,管壁增厚,由致密的炎症性纤维化组织组成,平滑肌稀少,有少量或没有上皮内膜覆盖。囊状扩张的体积不一,腔内液体可自数十毫升以至千余毫升。囊内胆汁的色泽取决于梗阻的程度,胆汁黏稠或清稀呈淡绿色,胆汁可以无菌,如合并感染,常为革兰阴性菌。炎性病变发展较突然者,甚至可引起管壁穿孔。可发现囊内有小粒色素结石存在。恶变率随年龄的增长而增加,小儿病例不足1%,而成人病例高达15%,病理组织学证明,以腺癌为多,在囊壁组织及免疫组织化学的研究中,发现胆管上皮化生与癌变相关。
胆管阻塞的持续时间决定肝脏的病理改变,在早期门脉系统炎性细胞浸润,轻度胆汁淤积和纤维化。在婴儿,胆管增生和小胆管内胆汁填塞,类似胆管闭锁所见,但病变是可逆性的。如果梗阻持续和(或)上行性胆管炎发生,则有胆汁性肝硬化,并可继发门静脉高压及其并发症,腹水及脾肿大也有所见。
本病一经确诊应尽早手术,否则可因反复发作胆管炎导致肝硬化、癌变或囊状扩张胆管破裂等严重并发症。完全切除扩张胆管和胆肠Roux-en-Y吻合是本病的主要治疗手段,疗效良好。完全切除扩张胆管困难时,可仅将扩张胆管黏膜完整剥离切除。对于并发严重感染或穿孔等病情危重者,可先采用胆汁引流术,待症状控制、黄疸消退、一般情况改善后,再行二期扩张胆管切除和胆肠内引流术。对于合并局限性肝内胆管扩张者,可同时行病变段肝切除术。如肝内胆管扩张病变累及全肝或已并发肝硬化,可考虑施行肝移植手术。









