


 收藏
收藏 已收藏
已收藏在医学上,性别可分为生理性别和心理性别。心理性别是后天形成的,形成期约在幼儿期3岁左右,是人对自我的认识。心理性别的异常主要是自我认知出现倒错导致的精神神经疾病,可伴或不伴生理性别的异常。生理性别是先天形成的,可分为染色体性别、性腺性别和表型性别3种类型。染色体性别是由父母双方的染色体正常构成及分裂决定的,男性为46,XY,女性为46,XX;性腺性别是由染色体决定的,男性为睾丸,女性为卵巢;表型性别的分化起源于同一始基,胎儿体内的激素是其性别分化方向的决定因素,在雄性激素的作用下分化为阴茎和阴囊,在缺乏雄性激素作用的情况下分化为阴蒂和阴唇。在正常情况下,3种性别类型是相一致的,通常所说的性别就是指表型性别,即外生殖器的性别。性腺性别与表型性别不符成为假两性畸形,表现为46,XX伴男性表型或46,XY伴女性表型。染色体性别与性腺性别不符称为性反转,表现为46,XY,内生殖器为卵巢,无睾丸及前列腺精囊;或是46,XX,内生殖器为睾丸,无卵巢及子宫输卵管。
先天性肾上腺增生症是一组常染色体隐性遗传疾病,发病率约1∶20 000。皮质醇激素合成过程中,关键酶活性的缺失可导致双侧肾上腺皮质增生伴原发性功能低下,并有性腺激素合成异常,导致性征异常。患者的父母是杂合子,患者为纯合子或复合杂合子。以21α-羟化酶缺陷症最常见(约占90%),11β-羟化酶缺陷症和3β-类固醇脱氢酶缺陷症(5%~8%)次之,而17α-羟化酶缺陷症和类固醇激素急性调节蛋白(steroid acute regulatory protein,StAR)缺陷症罕见。
1.病因及发病机制
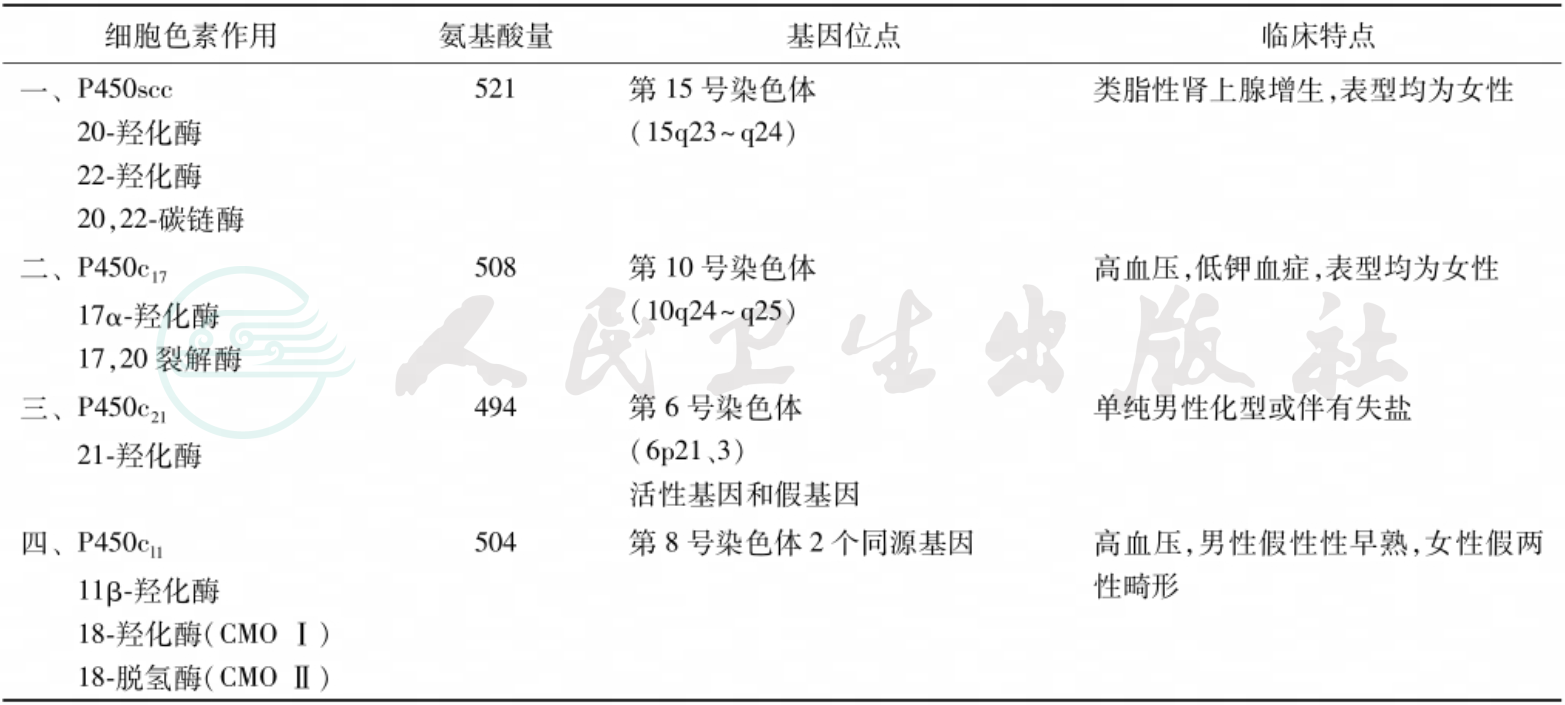
胆固醇通过类固醇羟化酶系统的电子传递即羟化过程,最终合成类固醇激素。细胞色素P450系列酶参与皮质醇激素合成的羟化过程,不同酶在肾上腺皮质不同区带中分布不同(表1)。P450酶的缺陷使皮质醇激素合成部分或完全受阻,下丘脑-垂体的CRH-ACTH分泌反馈性增加,刺激肾上腺皮质过度增生。此外,不同的P450酶缺陷对皮质醇以外的类固醇激素影响不同,导致不同的前体物质的蓄积,产生相应的临床和生化改变。
表1 各P450的特性及基因位点
2.分类
根据P450酶缺陷的类型,CAH可分为5型。突变的酶蛋白尚保留不同程度活性,故临床表现可从重型到非经典型(表2)。
表2 CAH分类及临床表现

(1)P450c21(21-羟化酶)缺陷
1)失盐型
2)单纯男性化型
3)迟发型(或成年发作型)
(2)P450c11(11β-羟化酶)缺陷
1)重型
2)迟发型(或成年发作型)
(3)P450c17(17α-羟化酶,17,20裂解酶)缺陷
(4)3β类固醇脱氢酶缺陷
1)重型
2)迟发型(或成年发作型)
(5)P450scc(20,22碳链酶,20和22羟化酶)缺陷
此外,18-羟化酶缺陷由于不影响皮质醇合成,对下丘脑-垂体有正常反馈,无ACTH分泌增多,所以无肾上腺皮质增生。
3.临床表现
(1)P450c21(21-羟化酶)缺陷
为先天性肾上腺皮质增生症中最为常见的类型,占总数90%~95%。
1)原发性肾上腺皮质功能减退
根据缺陷酶保留活性的不同,分为失盐型(重型)、单纯男性化型及迟发型(非经典型或成年发作型)。表现为不同程度的厌食、恶心、呕吐、低血糖等症状,重者出现低钠血症、高钾血症、代谢性酸中毒。ACTH增高使得患者出现类似艾迪生征的体征,即皮肤、黏膜色素沉着,在皮肤皱褶处如手指关节伸面、腋窝、腹股沟、乳晕周围尤为明显。
2)性分化异常
胚胎发育期即伴随的高雄激素血症,导致女性男性化或男性性早熟。女性外生殖器的男性化改变称“女性假两性畸形”,出现阴蒂增大,频繁勃起,性欲亢进,易有手淫习惯。但内生殖器仍为女性型,可有原发或继发性闭经、不育、多囊卵巢。男性表现为出生后1~2年外周性性早熟,外生殖器发育加速,3~4岁时阴茎、前列腺如青春期大小,常显示发育过度,即所谓“早熟性生殖器巨大畸形”。未经治疗的成年男性,其间质细胞功能、精子生成大多正常,少数患者没有正常青春期,睾丸体积小,出现无精子及不育。由于肾上腺皮质与性腺均来源于中胚层的体腔上皮,故在胚胎发育过程中,少部分肾上腺细胞随性腺迁移,可在性腺中形成肾上腺残余肿瘤。在治疗不规律的男性患者睾丸中多见,称为睾丸肾上腺残余肿瘤(testicular adrenal rest tumour,TRAT),多为良性,但会造成生精小管阻塞、生精障碍。
3)第二性征异常
女性2~3岁即有阴毛、腋毛,到青春期喉结粗,音调低沉,无乳房发育,体毛增多,类似男性。男性3~4岁时阴毛、腋毛生长,体毛增多,有痤疮。儿童早期生长加速,肌肉发达,肩距宽,皮肤粗糙,比同龄人高大,又由于雄激素的作用使骨骺提前融合(11~12岁已完全融合),最后身高低于同龄人,体形粗矮丑陋,最终长成矮小宽肩的小“大力士”体型。
4)性格改变
好动,注意力不集中,学习成绩差,可能与高雄激素有关。
(2)11β-羟化酶缺陷
此型少见,约为先天性肾上腺皮质增生的3%,可分为重型和迟发型。由于去氧皮质酮(DOC)过度蓄积并在外周组织转化成19-正去氧皮质酮(19-Nor-DOC),具有较强的潴钠作用,可部分代偿肾上腺糖皮质功能不足,掩盖盐皮质激素不足所致的失盐,可引起高血压,甚至为重度高血压、伴低钾血症。少数患者有较高的酶活性保留,DOC增加不明显,被醛固酮分泌减少而抵消,可表现为正常血压。这类患者临床上易与21-羟化酶缺乏单纯男性化型混淆。偶尔出现新生儿轻度失盐,但随着年龄增长,DOC逐渐增加,逐渐转为高血压、低钾血症。女性患者出生时外生殖器雄性化类似21-羟化酶缺陷,唯程度不如后者明显。迟发型往往在青春期发病,表现为多毛、痤疮、月经紊乱、不育,可有阴蒂肥大(无大阴唇融合),高血压可有或无,男性病儿往往难以识别,唯一诊断线索是快速生长和阴毛早现。
(3)17α-羟化酶缺陷
本征甚为罕见,多数患者在青春期因原发性闭经或青春期延迟而就诊。女性至青春期乳房不发育,无腋毛、阴毛,无月经,外阴幼女式、体型瘦高、肤色黝黑。男性(46,XY)由于胚胎期无睾酮,外生殖器似女性或部分男性化,往往作为女性培养。但无子宫、输卵管,睾丸位于腹股沟或腹腔内。在青春期往往有不同程度高血压,有的7~8岁即出现高血压,个别有严重高血压,一般抗高血压药难以起效。低钾血症多见,患者常伴有无力、疲劳、夜尿,甚至麻痹,骨骺融合延迟,停留在青春期。因被极高的皮质酮所代偿,故无肾上腺皮质功能减退表现。少数不典型病例可表现为正常血压或两性畸形。
(4)3β-羟类固醇脱氢酶缺陷
此型甚为少见。酶的缺陷同时累及肾上腺和性腺(卵巢、睾丸),男性胚胎不能分泌足够睾酮,致使出生时男性化不完全,有尿道下裂、隐睾,甚至男性假两性畸形。由于过量脱氢异雄酮(DHA)蓄积,可在周围组织部分转变成睾酮,女性可有轻度阴蒂肥大,大阴唇融合。婴儿皮肤黑与原发性肾上腺功能减退、ACTH增高有关。由于盐皮质激素和糖皮质激素均严重不足,出生后即有肾上腺皮质功能不足表现:厌食、恶心、呕吐、失钠及失水,最后因循环衰竭而死亡,即使及时诊断并治疗,多数仍难免夭折于儿童早期。轻型不典型3β-羟类固醇脱氢酶缺陷占本病10%~15%。与21-羟化酶缺陷迟发型类似,出生时无异常发现,发病多见于青春期,女性男性化类似21-羟化酶缺陷,是女性青春期多毛原因之一。由于DHA在周围转变成睾酮,男性至青春期有足够男性化,ACTH兴奋后,17羟Δ5孕烯醇酮、DHA明显增加,17羟Δ5孕烯醇酮/17羟黄体酮比值、17羟Δ5孕烯醇酮/皮质醇比值高于正常,据此可确诊。某些3β-羟类固醇脱氢酶缺陷者,血17-羟黄体酮甚高,接近21-羟化酶缺陷水平,这是由高浓度17-羟孕烯醇酮在周围转变而成。同样,因有明显女性男性化,使其与21-羟化酶缺乏诊断发生困难,唯一鉴别依据是17羟Δ5孕烯醇酮与17羟黄体酮的比值增高。
(5)P450scc缺陷(类脂性肾上腺增生)
由于胆固醇碳链酶(20,22-碳链裂解酶)、20-羟化酶或22-羟化缺陷,使胆固醇转变成孕烯醇酮受阻,因而糖、盐、性皮质激素均不能合成,无论染色体性别是男(XY)或女(XX),出生时外生殖器均为女性或近乎为女性。男性患者在腹股沟或腹腔内可找到睾丸。出生后立即出现肾上腺皮质功能减退及其危象,伴有明显皮肤色素沉着、厌食,恶心、呕吐,腹泻,体重丧失,脱水,尿17-OHCS、17-KS低下。由于缺乏糖皮质激素,免疫功能不全,感染多见,即使立即给予皮质醇和DOCA,亦多夭折于婴儿期。尸解见肾上腺增大,切面黄色、泡沫状,细胞内充满类脂质。
4.实验室和辅助检查
(1)常规、生化检查
1)21α-OHD和11β-OHD睾酮水平升高,可使红细胞增多,血黏度升高。CAH失盐型者有效血容量不足,HCT升高。
2)血、24小时尿同步电解质:StAR缺陷、3β-HSD、21α-OHD者可有低钠血症、高钾血症,尿路失盐,血钙升高,尿钙增加;11β-OHD、17α-OHD低钾血症、尿钾排出增多,血钙正常或偏低。
3)血气分析+同步尿可滴定酸:StAR缺陷、3β-HSD、21α-OHD可有代谢性酸中毒,尿液酸性;11β-OHD、17α-OHD可有代谢性碱中毒,尿液碱性。
(2)内分泌激素检查
1)CAH可伴有糖代谢异常,表现为高胰岛素血症、IGT、糖尿病。
2)ACTH:StAR缺陷、3β-HSD、21α-OHD失盐型明显升高,可>1000ng/L,3β-HSD非经典型、21α-OHD单纯男性化型和非经典型、11β-OHD、17α-OHD可升高,但根据酶活性,ACTH范围介于正常高限至1000ng/L之间。ACTH呈脉冲分泌,应在同一时间段或不同时间段重复检查至少2次。
3)血F节律、24小时尿皮质醇:CAH经典型患者血、24小时尿F明显降低,非经典型者血、尿F可降低、正常,甚至轻度升高,但相对于升高的ACTH,仍显分泌不足。
4)性激素:StAR缺陷FSH、LH升高,所有性激素均低下;3β-HSD者FSH、LH正常或偏低,DHEA升高,余性激素均低下,但非经典患者高浓度的DHEA和17-羟孕烯醇酮可在外周转化,引起T和17-OHP升高;21α-OHD和11β-OHD的FSH、LH正常或偏低,余性激素均升高,17-OHP明显升高,>10μg/L,孕期羊水17-OHP检测亦可用于21α-OHD的胎儿筛查;17α-OHD的FSH、LH、P明显升高,其他性激素均明显低下。
5)21α-OHD、11β-OHD、3β-HSD非经典型雄激素升高,PTH、骨密度25(OH)维生素D、骨钙素均可正常,StAR缺陷、3β-HSD经典型和17α-OHD性激素不足,钙吸收不良,PTH、骨钙素升高,骨密度、25(OH)维生素D降低。
(3)动态试验
1)24小时动态血压监测
17α-OHD、11β-OHD可伴血压中重度升高,表现为难治性高血压;21α-OHD非经典型长期高雄激素血症可导致代谢综合征,亦可出现血压轻、中度升高。
2)快速ACTH兴奋试验
用以检测肾上腺皮质储备功能和排除CAH特别是21α-OHD。推注ACTH后1小时血F<18μg/dl,可确诊肾上腺皮质功能不足;17-OHP<10μg/L,可排除21α-OHD;P明显升高(>基础2倍)而17-OHP低于正常,可确诊17α-OHD;DHEA、AD高于正常值者,可认为肾上腺来源雄激素过多。
3)地塞米松试验
地塞米松2mg,分次口服7天,服药前后测定ACTH、血F(8am)、T、P、DHEA、AD和血钾,CAH等指标均恢复正常。
(4)特殊检查
1)21α-OHD、11β-OHD、3β-HSD非经典型骨龄>实际年龄,StAR缺陷、3β-HSD经典型和17α-OHD骨龄延迟。
2)17α-OHD、11β-OHD因高血压、低钾血症可见u波,心率增快,失盐、脱水者亦可有心率增快。
3)性腺超声:46,XY男性患者21α-OHD、11β-OHD、3β-HSD非经典型者睾丸体积偏小、正常或增大,StAR缺陷、17α-OHD男性假两性畸形者睾丸可能位于腹腔、腹股沟、大阴唇,亦有可能已经萎缩,无法找到。46,XX女性患者21α-OHD、11β-OHD、3β-HSD非经典型者可有多囊卵巢结构,子宫小、内膜薄;StAR缺陷、17α-OHD可有幼稚子宫、条索状卵巢表现。
4)肾上腺CT:双侧肾上腺明显均匀性增生,21α-OHD、11β-OHD以网状带增生明显,严重者可表现为腺瘤样增生,因网状带靠近髓质,有时亦累及髓质增生;17α-OHD因脱氧皮质醇的代偿作用,肾上腺轻度增生。
5)21α-OHD、11β-OHD者可伴有少精子症、无精子症,非经典型精液正常。
6)染色体核型分析:21α-OHD、11β-OHD可伴有女性假两性畸形,StAR缺陷、17α-OHD伴有男性假两性畸形,3β-HSD表型性别模糊,均需确定染色体性别。
7)基因诊断:对于临床难以确诊的CAH,可行基因筛查以排除;临床诊断为CAH患者,可通过基因诊断进一步分型,并可行家系筛查确认患者和携带者,对有再次生育要求的配偶可行妊娠期羊水脱落细胞培养检查或胎盘绒毛膜活检细胞基因诊断。
5.诊断
CAH各型的临床表现不一,患者常因肾上腺外症状就诊,尤其是非经典型患者极易误诊和漏诊。StAR缺陷和3β-HSD非常罕见,且肾上腺皮质功能减退病情较重,同21α-OHD失盐型患者一样,出生时即有症状可确诊。而对于21α-OHD非经典型、11β-OHD和17α-OHD缺陷早期症状不明显,往往在青春期年龄起病,是成人内分泌科重点关注的CAH类型。凡有青少年起病的原发性肾上腺皮质功能低下伴双侧肾上腺增生合并高血压、月经失调、多毛、皮肤色素沉着、性早熟、外阴畸形、失盐、脱水、电解质紊乱等症状之一的,均要考虑本病。怀疑本病者,应详细询问母亲妊娠史、家族史、出生史、哺乳史、生长发育史、月经史。测量身高、体重、上部量、双臂展长度,进行毛发评分、性腺及第二性征评估。
6.鉴别诊断
(1)“呆小病”:先天性腺垂体功能低下者有甲状腺功能减退,CAH患者甲状腺功能正常。
(2)StAR缺陷:3β-HSD、21α-OHD者肾素明显升高,醛固酮降低、正常或升高,但血压正常或偏低,可与高血压伴继发性醛固酮增多症鉴别,应与醛固酮不敏感综合征鉴别。
(3)21α-OHD:常误诊为髓样脂肪瘤或肾上腺腺瘤;11β-OHD、17α-OHD者有高血压、低钾血症,且肾素明显降低,醛固酮降低,ARR比值>300,常被误诊为原发性醛固酮瘤或嗜铬细胞瘤而进行不必要的手术,术后病情加重。因此,CAH需与双侧肾上腺增生性疾病如库欣病、异位ACTH增多症、大结节增生、原发性色素沉着性小结节样增生、增生性原醛、嗜铬细胞瘤、髓样脂肪瘤、纤维性骨营养不良综合征、肾上腺皮质癌、肾上腺淋巴瘤、肾上腺结核、双侧肾上腺腺瘤等仔细鉴别。
(4)CAH者垂体正常或饱满,可与先天性下丘脑-垂体发育不良、垂体ACTH瘤或颅咽管瘤、生殖细胞瘤、朗格汉斯细胞组织细胞增生症、自身免疫垂体炎等鉴别。
(5)CAH患者的HCG正常,可与肿瘤性疾病HCG升高导致性腺发育异常鉴别。
(6)CAH伴矮小者GH正常或升高,IGF-1、IGF-BP3正常,应与生长激素缺乏所致矮小者鉴别。
(7)染色体疾病如特纳综合征、克兰费尔特综合征,大或小Y染色体,染色体异位、缺失等均有可能导致性腺发育异常,需要染色体分析与CAH进行鉴别。
7.治疗
早期诊断、治疗甚为重要,特别是21α-OHD和11β-OHD缺乏,如诊治始于胚胎早期,可阻止雄性化出现,获得正常发育婴儿;如出生时未能识别,常导致后期发育异常,严重病例则夭折于婴儿期。
(1)肾上腺皮质激素替代治疗(抑制性替代),皮质醇或可的松为首选药物,维持剂量的大小因人而异。
(2)对于有失盐表现的患者特别是婴幼儿,应及时加用盐皮质激素,随着年龄的增加,机体理盐功能逐步完善后可停用。
(3)对于伴有高血压、低钾血症的CAH患者,在糖皮质激素治疗起效之前辅以ACEI、ARB类降压药或螺内酯治疗。
(4)对于伴有雄激素过高、女性男性化或男性性早熟患者应阻止雄性化,使生长速率减慢,骨骺融合接近正常年龄,尽可能达到正常身高,恢复正常排卵和生育能力。
1)促黄体生成素释放激素(LHRH)类似物和生长激素(GH)治疗可有效抑制CAH患者的真性性早熟,改善最终身高。
2)女性患者可辅以雄激素受体拮抗剂氟他胺治疗。
3)芳香化酶抑制剂如来曲唑可减少雄激素向雌激素转化,防止骨骺过早融合。
4)伴有多囊卵巢、胰岛素抵抗者,可加用胰岛素增敏剂。
5)手术整形治疗:女性患者伴有阴蒂肥大、阴唇融合等雄性化表现者,应切除过多部分的阴蒂,做阴道成形术。乳房发育不良者,必要时可行隆乳术。
(5)对于同时伴有性激素合成缺陷的CAH患者,表现为女性性幼稚、男性假两性畸形,临床上均为幼女外观,治疗应以改善性征为主。
1)雌激素替代治疗,以促使乳房和外生殖器发育。同时,还可用适量睾酮刺激阴毛生长。
2)手术整形治疗:乳房发育不良者,可行隆乳术。如表型女性,而遗传性别是男性(46,XY)有隐睾者,其治疗应根据社会性别决定,因隐睾恶变可能性较大,应予尽早切除。
(6)要对患者及其家长强调治疗的重要性,女性必须终生治疗,遇应激时应加大治疗剂量,否则可导致致命的肾上腺皮质功能不足危象。
(7)其他:CRH拮抗剂以减少ACTH生成、基因治疗等。
由于编码雄激素受体(androgen receptor,AR)的基因突变致AR结构和功能异常,造成男性性发育有关的靶组织对雄激素不敏感,从而使雄激素的正常生物学效应部分或全部丧失,引起男性假两性畸形。
1.临床表现
该病为X染色体隐性遗传,根据患者不同程度的临床表现可分为完全性AIS、不完全性(或部分性)AIS及轻度AIS。
根据患者有无男性化表现,将AIS患者分为无男性化表现的完全型和有男性化表现的不完全型2大类。完全型雄激素不敏感患者由于在胚胎期雄激素作用的完全缺失,造成表观性别发育为女性型,所以自幼均按女性生活,成年后临床表现为原发闭经,由于完全缺乏雄激素的抑制,少量的雌激素即可导致乳房发育和女性体态,但乳头发育差,阴、腋毛无或稀少,女性外阴,大、小阴唇发育较差,阴道呈盲端,无宫颈和子宫,人工周期无月经。不完全型雄激素不敏感患者的临床表现范围变化极大,与完全型的主要区别在于有不同程度的男性化。
2.辅助检查
(1)染色体分析
AIS患者染色体为46,XY。
(2)测定体内性激素水平
AIS患者体内雄激素高于女性正常标准。
(3)SHBG雄激素敏感试验
可反映雄激素受体缺陷严重程度。给予司坦唑醇0.2mg/(kg·d)每晚顿服,连服3天。分别在服药前和服药后第5、6、7、8天抽血测SHBG。刺激后SHBG最低值与刺激前浓度的百分比值作为指标,完全性雄激素抵抗者均值为102.2%±2.1%。
(4)性腺超声
无子宫样回声,腹股沟区可见睾丸样回声。
(5)分子诊断
对于可疑患者,可直接提取外周血白细胞DNA进行基因诊断。常用PCR-SS-CP、等位基因特异性PCR、外显子CAG重复序列长度分析等方法检测AR基因的结构异常。
3.诊断
染色体检查结果为46,XY,出生和儿童时期表现为女性并在腹股沟区或阴唇内触及睾丸样组织,青春期后出现女性第二性征,原发性闭经,腹股沟或阴唇内肿物等典型临床表现可确诊。
4.治疗
治疗的目的为确定社会性别、矫正畸形,并尽可能维持性功能和消除心理障碍。对于位于大阴唇或腹股沟管的睾丸,应在青春期后切除以获得自发性乳房发育。对于位于腹腔内的睾丸,目前认为应尽早切除以预防睾丸肿瘤。轻度男性化的患者应在青春期前或刚启动时切除睾丸,以防止男性化加重。睾丸切除后应予雌激素替代治疗,以促进第二性征发育。同时,对患者予以心理疏导,除了不育之外,患者可经历完全正常的女性生活。
5α-还原酶位于细胞内质网质膜上,主要作用为催化睾酮转变为活性更强的双氢酮。此外,也以其他类固醇激素如脱氢表雄酮、黄体酮、氢化可的松等作为底物,催化其代谢过程。
1.临床表现
5α-还原酶缺乏的典型临床表现为出生时两性畸形,类似阴蒂肥大,尿道下裂。睾丸分化良好,位于腹股沟管或阴唇阴囊褶内,附睾、输精管、精囊腺分化良好。无子宫及输卵管。射精管常开口于阴道盲端。前列腺缺如或发育不良。多数患者出生时作为女性抚养,青春期患者出现显著男性化:声音低沉,肌肉增加;阴茎增长,常伴有不同程度的痛性勃起,甚至射精;外阴皮肤色素沉着但阴毛腋毛缺如;睾丸增大。无乳腺发育,前列腺不发育。
2.诊断
(1)T/DHT比值
正常成年男性外周血T/DHT的比值为12±3.1,而成年5α-还原酶缺陷症患者比值可达35~84。青春期前的患者可通过HCG兴奋试验诱发T/DHT比值的异常。
(2)5α-还原酶活性测定
是确诊5α-还原酶缺陷症的直接手段。方法为将患者外阴活检皮肤标本加入放射性标记的睾酮作为底物,测定作为产物的DHT的含量,从而测定5α-还原酶的活性。
(3)基因检测
对SRD5A2基因直接进行分子遗传学检测。
3.治疗
应强调早期诊断,以便尽早决定抚养性别,避免青春期改变性别对患者心理造成影响。性别的选择主要取决于外阴发育情况,男性化缺陷严重者应该按照女性抚养,青春期前应切除睾丸,并在青春期年龄进行雌孕激素周期替代治疗,必要时行阴道成形术。选择按照男性抚养者,可行外生殖器整形术,如尿道下裂修补术、阴茎尿道成形及睾丸固定术,同时给予雄激素替代治疗。目前,可用于替代治疗的雄激素有双氢睾酮和睾酮。对于患者的生精功能障碍尚无治疗方法。
真两性畸形的定义是体内同时存在睾丸和卵巢两种性腺组织。染色体核型最多为46,XX,其次为46,XX/46,XY嵌合,极少为46,XY。临床表现为外生殖器两性畸形,尿道下裂,阴囊阴唇褶部分融合,多数伴有隐睾,但是至少有一侧的性腺可在腹股沟区或阴囊阴唇褶内触及。多数患者具有子宫和阴道,子宫可发育不良,少数患者子宫缺如。最常见的性腺类型是卵睾(在一个性腺内同时存在睾丸和卵巢两种组织),其次为卵巢,睾丸最少见。卵睾和卵巢一侧的生殖导管为输卵管和子宫,睾丸一侧为输精管。青春期后常有乳腺发育,半数以上患者可有月经来潮。46,XX核型患者性腺一侧为卵巢时,卵巢功能可正常,可有排卵、受孕和生育。两性腺一侧为睾丸时,生精小管发育不良,精子发生少见。治疗上应依据患者的年龄和内外生殖器情况评估而定。在患者尚无性别认同的时期,46,XX核型的患者除非有发育良好的阴茎或子宫严重发育不良,均应按照女性来抚养,并切除睾丸组织,行外生殖器整形术,如果青春期后卵巢功能不足,可予人工周期治疗。如果按照男性抚养,需要切除卵巢、子宫和输卵管,由于睾丸和卵睾中的睾丸组织发育不良,易发生肿瘤,因此应行预防性性腺切除术及外生殖器整形术,青春期时给予雄激素替代治疗;46,XX/46,XY或46,XY核型的患者,宜作为男性抚养,但应警惕性腺发生恶变可能。对于年龄较大的患者,性别取向应以患者认同的社会性别为依据,切除与性别不一致的性腺组织,行相应的外生殖器整形术,青春期后给予相应的性激素替代治疗。









