


 收藏
收藏 已收藏
已收藏朗格汉斯细胞组织细胞增生症(Langerhans cell histiocytosis,LCH)曾称为组织细胞增生症X(histiocytosis X),是一种非肿瘤性朗格汉斯细胞异常增生或反应性增殖性疾病,此种增生可能是由于内源性或外源性刺激导致免疫调节功能紊乱所致。1953年,Lichtenstein对其进行了经典分型:莱特勒-西韦病(勒-雪病,Letterer-Siwe disease,LSD),汉-许-克病(韩-薛-柯病,Hand-Schüller-Christian syndrome,HSC),骨嗜酸性粒细胞肉芽肿(eosinophilia granuloma of bone, EGB)。1973年,Nezeloff在电镜下观察到其细胞内有与皮肤朗格汉斯细胞相同的Birbeck颗粒,以后免疫组织化学技术的发展证明了其细胞和朗格汉斯细胞及其他染色体T区组织细胞具有共同抗原,并具有幼淋巴细胞存在的T4抗原,其他T区组织细胞所不具有的Birbeck颗粒,提示其为朗格汉斯细胞起源的肿瘤性或反应性增生性病变,故1985年全美组织细胞学会建议应用LCH取代旧的命名。
儿童LCH的发病率约为2~5/100万,男性略高,发病高峰在1~4岁。本院临床资料显示1984 年5月~2000年9月的16年间,收治约42例LCH,男25例,女17例,男女之比为1.5∶1,年龄8个月~13岁,中位年龄为3岁。估计有相当一批患者以EGB就诊于小儿外科或口腔科。
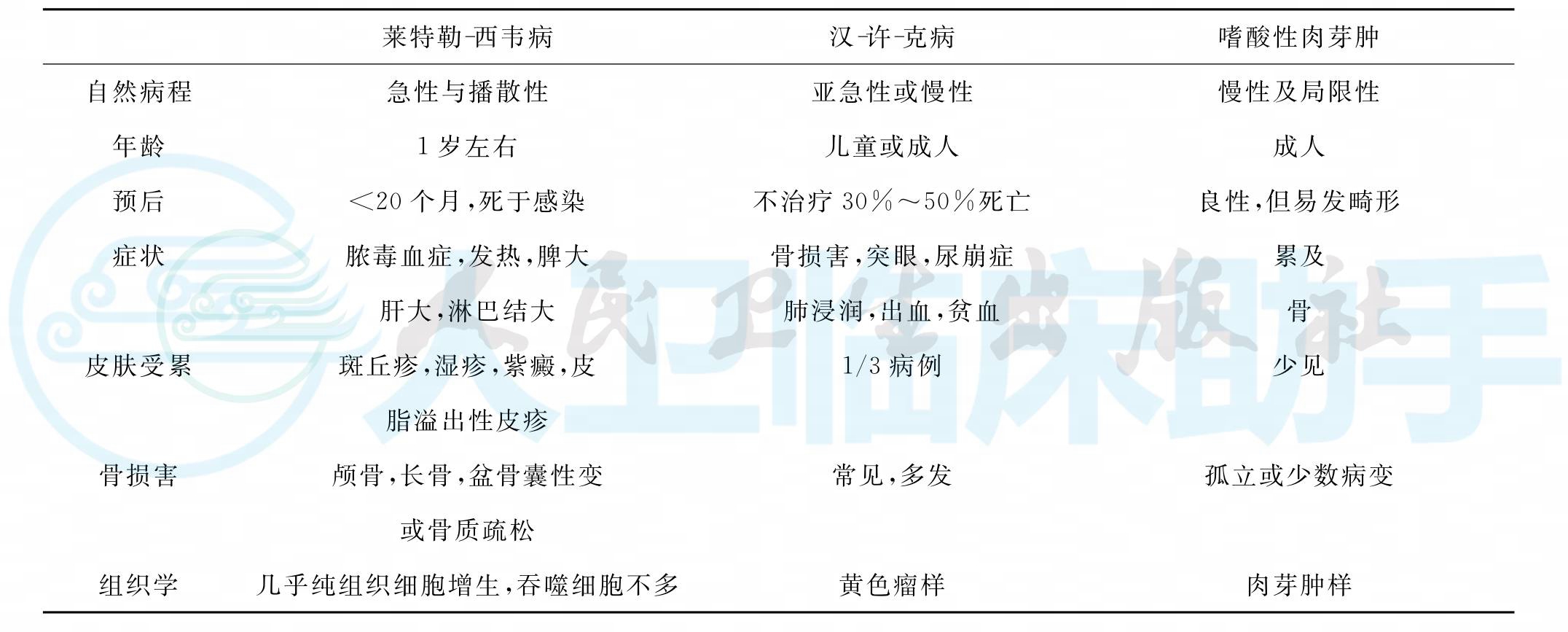
LCH的临床经过迥异,LSD在婴幼儿期发病,表现重,易发生多脏器功能损害及衰竭,合并感染,进展迅速,病死率高。HSC在幼儿和学龄前儿童发病,以膜性骨的溶骨性损害、突眼、尿崩症为特点,虽无生命危险,但呈慢性进展,迁延难治。EGB成人多见,而儿童在年长儿发病侵犯长骨,也见于颅骨、椎骨、肋骨、骨盆骨等扁骨,可呈单一性或多发性病灶,预后多良好,但也有骨折、跛行、肢体功能障碍等并发症(表1)。
表1 三种朗格汉斯细胞组织细胞增生症特点比较
随着近年来分子生物学及细胞遗传学的进展,大量的基础研究发现:
1.LCH可能属肿瘤性克罗恩病,因细胞突变导致骨髓和其他器官中LC或其前体呈克隆性增殖。
2.细胞因子介导性疾病,LCH的受累组织中某些细胞因子水平升高,G-CSF和TNF-α共同作用,促使造血干细胞(CD34前体细胞)产生LC(20%含有Birbeck颗粒),故推断LCH的LC增殖与该两种因子受到刺激有关。
3.感染可能引起LCH的增殖,如HSV-6感染后的反应过程。
患儿,女,10个月,因“头部反复性皮疹5个月,发热1个月”来诊。5个月中,头皮、前胸、后背、腋下分批出现湿疹样皮损,有渗出及结痂。近1个月持续性不规则高热(37.5~39.8℃),双耳流脓,久治不愈。黑绿色稀水样便,每天3~10次。院外曾用地塞米松治疗,热退又复现,间断应用头孢类抗生素达1个月。平素体弱,易患“上感”。家族史:母亲39岁,父亲29岁,其胞姐16岁,健康。
体格检查:T 39.8℃,BW 10kg。神志清,营养中等。方颅,头皮、眉弓、双耳、耳后可见湿疹样皮损,局部连成片,眉弓破溃处结痂。浅表淋巴结肿大,呈黄豆大小,无压痛,活动好。前胸数个白斑,右牙槽区0.5cm×0.5cm溃疡,右下颌肿大区3cm× 4cm,无压痛与骨相连。咽部充血,双扁桃体不大,双耳道有少量黄色分泌物,颈软,呼吸音粗,腹平软,肝大,肋下3.0cm,脾肋下3.5cm,均Ⅱ°。
实验室检查:①血常规:WBC 12.1×109/L,N 0.34,L 0.63,Hb 93g/L,RBC 4.08×1012/L,PLT 187×109/L,MCV 72fl,嗜酸性粒细胞计数66× 109/L。②尿常规:比重1.020,蛋白阴性,pH 6.0, RBC 0~1/HP,WBC 0~2/HP。③便常规:WBC 8~10个/HP,稀黄便。肠道菌群:中度失调,真菌生长为光滑念珠菌。④血细菌培养2次阴性,真菌培养2次阴性。便培养生长光滑念珠菌,细菌培养阴性,用氟康唑后5天转阴。⑤ESR 35mm,CRP阳性,ASO阴性。肝功能:T 55.8g/L,A 38.0g/L, ALT 12U/L,TBiL 19.7μmol/L,直接胆红素16.3μmol/L,AST 61.8u/L,γ-GT 15U/L。肾功能:BUN 2.44mmol/L,Cr 87μmol/L。心肌酶谱: LDH 396U/L,CK 1323U/L,CK-MB 64U/L,α-HBDH 306U/L。⑥IgG 3.45g/L,IgA 0.09g/L,IgM 0.37g/L。T细胞亚群:CD3 0.86,CD4 0.56, CD8 0.20,CD4/CD8=2.8。⑦病毒IgM均阴性(EBV、RSV、ADV、CMV等)。HIV阴性,梅毒血清TPHA阴性。⑧胸部X线片:肺纹理增强,呈弥漫网状影,夹杂大小不等的囊状透光区。颅骨正侧位片:右下颌骨骨质肿物。B超:肝大,肋下约4.6cm,剑突下4.2cm,表面光滑,缘锐利,实质回声均匀;脾大,肋间厚3.0cm,肋下1.4cm,无积液及包块。全身骨骼ECT:TC99m显影清晰,无放射性浓聚现象。头颅CT:右下颌角处一圆形密度均匀的软组织影,侵蚀下颌骨质,边缘清楚,骨皮质略呈膨胀改变,周围软组织肿胀。⑨骨髓象:增生明显活跃,粒红比1.39∶1,粒系核左移,红系增生显著,部分可见巨幼变,巨核细胞44个。片尾可见类戈谢样组织细胞,形态特殊,胞体40~60μm,多为长形,胞质丰富,边缘模糊,染淡蓝灰、紫色,内含颗粒或吞噬物,可见空泡形成,染色质疏松,可见双核。头皮皮疹印片:单个核的组织细胞成堆可见,体积较大,染色浅,核椭圆或圆形,偶见核分裂象。
入院诊断:朗格汉斯细胞组织细胞增生症、莱特勒-西韦病、营养性缺铁性贫血(轻度)、光滑念珠菌性肠炎。
1.化疗 应用VP方案,长春新碱(VCR)0.6mg/次,每周1次;泼尼松10mg/d,连用5周。次日体温下降到36.2℃,但2天后又上升到40℃,呈不规则高热。
2.因便培养生长真菌,予氟康唑(Diflucan),50mg/d,用3天后停用3天,复查肝、肾功能正常,再用4天。
3.支持疗法
(1)营养支持:由于进食差,仅吮母乳,间断滴注脂肪乳剂及氨基酸。
(2)胸腺肽:5mg/d,连用1个月。
(3)增强体液免疫功能:丙种球蛋白2.5g/次,间断用9次。
(4)血液支持:VP化疗后因Hb下降到76g/L, PLT 62×109/L,输100ml全血。
4.局部处理,住院中头、颈、手指掌侧多个白色水疱样疱疹,考虑为皮肤霉菌病,外涂孔雀绿、霉克霜。
5.合并缺铁性贫血,口服血红素铁(益气维血颗粒)。
6.约4周后热下降到37℃左右,皮疹及肝脾大消失。血常规:WBC 11.5×109/L,Hb 102g/L, PLT 110×109/L。在出院随访中。









