


 收藏
收藏 已收藏
已收藏脊髓性肌萎缩(spinal muscular atrophy,SMA)是一种少见的常染色体隐性遗传病,以脑干运动核和脊髓前角细胞进行性丢失为主要特征。依据发病年龄和进展速度通常将SMA分为三型:SAMⅠ、SAMⅡ和SAMⅢ。婴儿型SMA(SMAⅠ)可发生于新生儿期,患病率为0.1/10万~1/10万,是导致肌张力低下症的常见运动单位疾病。
SMA发病机制是程序性细胞死亡缺陷,即在妊娠期间属正常的细胞凋亡过程延续到生后,使患儿生后运动神经元不断变性死亡,可于出生至成年任何时间出现症状。本病的致病基因是位于染色体5q12.2-q13.3的活运动神经元(survival motor neuron)基因SMN1。正常个体存在SMN1和SMN2基因,后者是位于同一染色体上几乎与SMN1相同的基因拷贝。SMAⅠ常归因于SMN2基因拷贝数的异常。近来研究表明,约98.6%的SMAⅠ患儿发病与该基因的缺失突变有关。SMAⅠ多在生后6个月以内发病,为进行性肢体无力,这种肌无力为对称性弛缓性软瘫,肌电图和肌活检为神经源性损害,为最有用的实验室诊断。本病的死亡率、病残率相当高,预后极差,目前尚无有效的治疗手段。
患儿,男,22天,以“四肢无力22天,呼吸困难5天”为主诉入院。患儿母系孕1产1,母孕40周,因脐带绕颈2周行剖宫产。母孕晚期胎动减少,出生时无窒息史。患儿出生后四肢无力,肢体活动少,尤以双下肢明显,但哭声尚正常,2~3天后声音逐渐减低,吸吮无力,自行哺乳差,入院前5天出现呼吸困难,并逐渐加重。父母非近亲结婚,身体健康,否认遗传代谢病史,家族中无类似病史。
入院查体:T 36.3℃,P 130次/min,R 40次/min, Bp 65/40mmHg,神志清,反应可,哭声弱,口唇发绀,鼻扇及三凹征阳性,双肺呼吸音粗,散在痰鸣音,未闻及湿啰音,心音有力,心率130次/min,节律规整,未闻及杂音,腹平软,肝脾不大,肠鸣音4~5次/ 分,双下肢无水肿,左足六趾畸形。神经系统查体:无面瘫,无眼球活动受限和眼睑下垂,痛觉存在,四肢肌张力明显减低,双上肢肌力Ⅱ级,双下肢肌力Ⅰ级,觅食、吸吮及吞咽均减弱,握持反射及拥抱反射消失。双侧PSR及ASR消失,双侧巴氏征(-)。辅助检查:血常规、C-反应蛋白、肝功能、肾功能及心肌酶谱等检查结果均正常;血清TORCH-IgM(-), 尿CMV-DNA(-);腰椎穿刺CSF常规及生化结果均未见异常;染色体核型分析46X,Y;血浆和尿液的GS/MS检查未见异常;胸片显示肺纹理增强;脊髓MRI扫描结果显示从颈段到胸腰段均未见异常(图1);头部MRI扫描:双侧颞部脑外间隙轻度扩大,脑实质未见异常(图2);肌电图检查显示:双胫前肌呈神经源性损害,右腓浅神经感觉神经传导,诱发电位未引出。
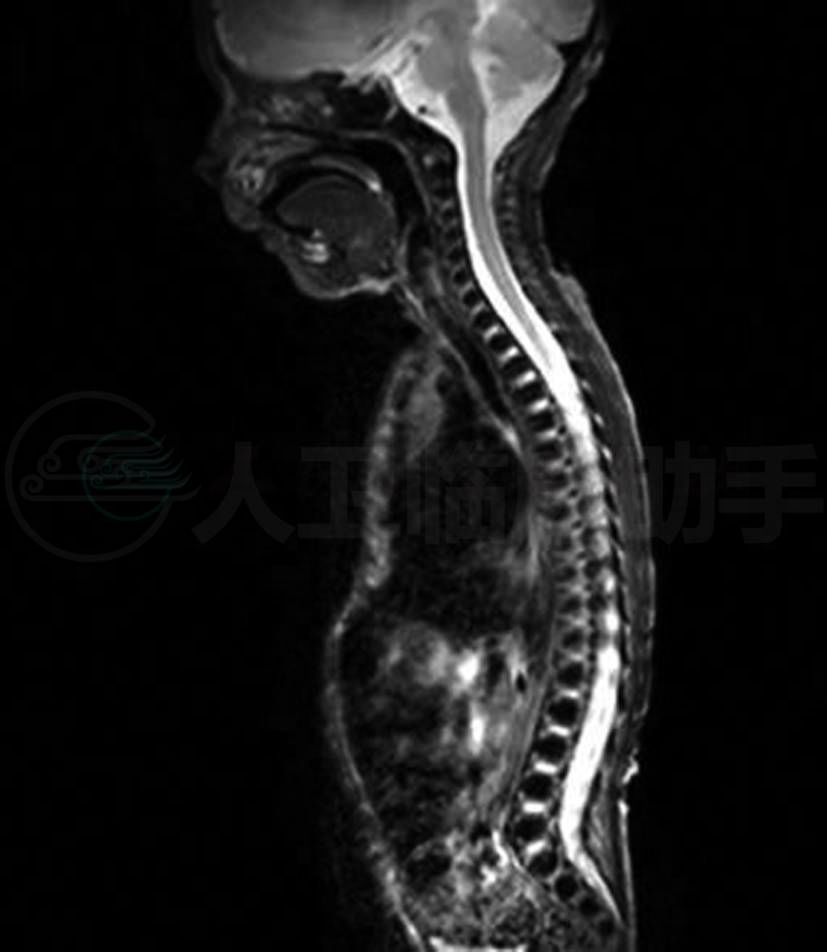
图1 脊髓MRI(T2)
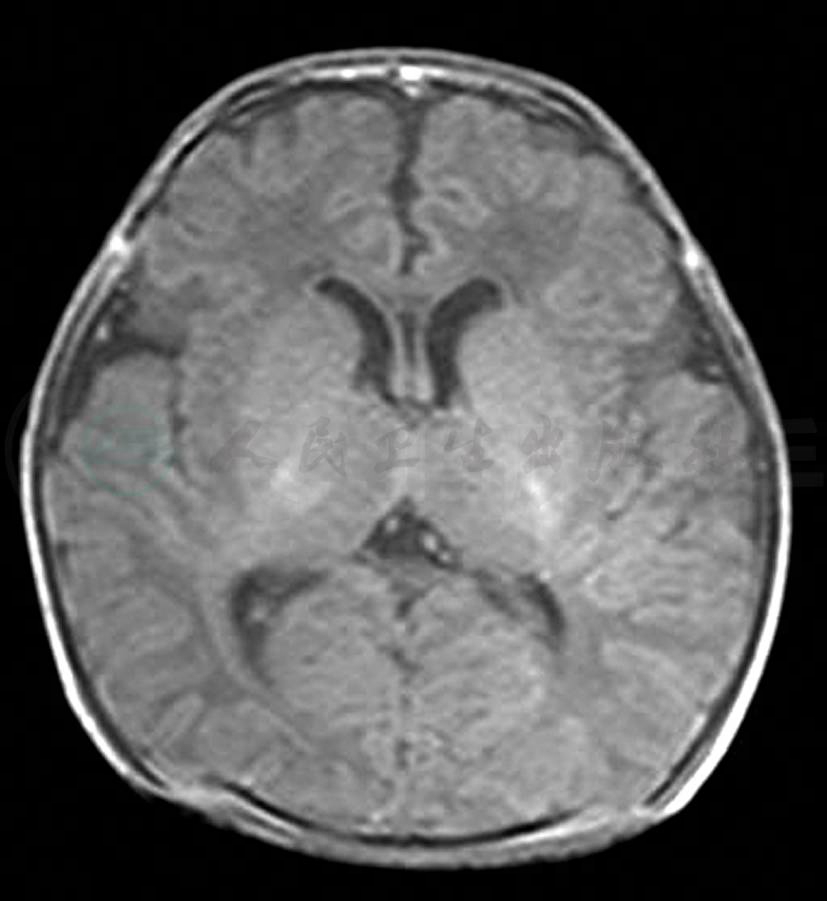
图2 头MRI(T1)
入院诊断:先天性肌迟缓综合征。
1.入院后保温、吸氧、抗感染等对症治疗。
2.住院期间,患儿病情逐渐进展,表现为哭声消失、喂养困难(需依靠胃管喂养)、呼吸浅表并出现胸腹式矛盾呼吸、肌张力消失、肌力下降(双上肢肌力Ⅰ级,双下肢肌力0级)。为进一步明确导致患儿神经源性肌损害的病因,对其进行了有关SMN基因缺失检测,结果发现患儿SMN基因第7外显子纯合缺失(图3)。
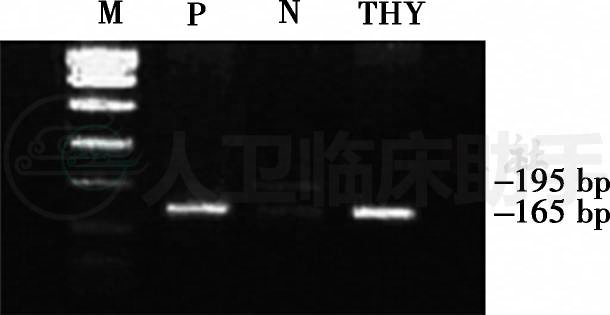
图3 PCR电泳图
注:M,marker;P,阳性对照;N,阴性对照;THY,患儿;PCR产物经DraⅠ酶切后仅见一条165bp电泳带,无190bp产物(SMN基因第7外显子)
最终诊断:婴儿型脊髓性肌萎缩症。









