


 收藏
收藏 已收藏
已收藏【主诉】
双下肢无力20个月。
【现病史】
患者,男性,43岁,河南省平顶山市居民。患者于入院前20个月(2008年1月)无诱因出现双下肢无力,远端为主,左侧较重,能自行走动,无疼痛和麻木等症状。当地以“低钾血症”予口服补钾治疗无效。16个月前(2008年5月25日),患者午睡后出现四肢不能活动,至医院“补钾”治疗后,症状好转,但双下肢无力症状较前加重,右侧为重,不能自行走动。13个月前(2008年8月)在当地医院就诊,1个月内3次肌电图均示:“广泛神经源性损害”。脑脊液及颈、胸椎MRI、血生化等“未见异常”。以“多发性周围神经病”给予“甲钴胺、鞘内注射地塞米松、神经生长因子”治疗,症状好转。5个月前(2009年4月)患者无诱因出现发作性双下肢肌肉酸痛、痉挛,渐发展至双上肢及颈部肌肉,于当地医院行新斯的明试验结果为“阳性”。诊断“重症肌无力”,予溴吡斯的明治疗,症状明显缓解。现患者可以行走,但双下肢无力致行走距离短。为进一步治疗至我院门诊,查肌电图:“神经源性伴神经性受损”;重复神经刺激:“右侧副神经、腋神经低频刺激时波幅出现递减现象(低频下降超过15%),高频刺激时波幅未见明显递增递减现象”。门诊医生以“重症肌无力?周围神经损害?”收住院。发病以来,无发热,精神可,二便无异常。发病以来体重减轻10余斤。
【过去史、个人史及家族史】
均无特殊。
【查体】
体温36. 2℃,脉搏76次/分,呼吸20次/分,血压125/85mmHg。内科系统检查未见异常。意识清楚,语言正常。智力、记忆力、定向力、计算力正常。脑神经检查未见异常。运动系统:双上肢肌容积正常,肌力5-级,肌张力正常。双下肢股四头肌、腓肠肌肌容积减小,骼腰肌肌力5-级,股四头肌肌力4级,胫前肌肌力1级,长伸肌肌力0级。双下肢肌张力低。感觉系统:双侧肢体痛触觉、振动觉、位置觉、运动觉对称正常。双侧肢体轮替试验、指鼻试验、跟膝胫试验稳准,Romberg试验正常。无不自主运动。蹒跚步态。双侧肱二、三头肌腱反射正常、左侧桡骨膜反射正常,右侧桡骨膜反射亢进。双侧膝腱反射未引出,跟腱反射减弱。双侧腹壁反射对称存在。吸吮反射、强握反射、掌颏反射阴性,Hoffmann征可疑阳性,双侧Babinski征、Chaddock征、Oppenheim征阴性、Gordon征阴性。颈无抵抗,Kernig征、Brudzinski征阴性。
【辅助检查】
1.2009年11月2日,11月25日,12月9日三次血生化结果 丙氨酸转氨酶59IU/L,61IU/L,49IU/L(0~40)。天门冬氨酸转氨酶45IU/L,13IU/L,18IU/L(0~37)。肌酸激酶1425IU/L,84IU/L(24~195)。肌酸激酶同工酶75IU/L,25IU/L,(0~25)。乳酸脱氢酶249IU/L,171IU/L,(109~245)。α-羟丁酸脱氢酶191IU/L,135IU/L,(76~195)。肌红蛋白,170. 6g/L,38g/L。
2.甘油三酯3. 51mmol/L(0. 4~1. 8)。总胆固醇6. 97mmol/L(3. 4~6. 7)。低密度脂蛋白胆固醇4. 12mmol/L(1. 9~3. 8)。高密度脂蛋白胆固醇1. 40mmol/L(0. 8~2)。
3.蛋白电泳 清蛋白59. 1%(52. 0%~62. 8%),α1球蛋白3. 5(3. 1~4. 6),α2球蛋白11. 9(7. 0~11. 1),β1球蛋白6. 2(5. 3~7. 8),β2球蛋白25. 2(3. 3~6. 4),γ球蛋白14. 1 (13. 1~23. 3)。
4.血清抗乙酰胆碱受体抗体阴性。
5.脑脊液检查 初压为200mmH2O,末压为100mmH2O。脑脊液常规:透明,浅红色,红细胞,圆盘状。细胞总数17 820×106/L。白细胞数8×106/L。蛋白定性试验(Pandy试验) (++)。蛋白0. 83g/L。糖3. 95mmol/L。氯化物124mmol/L。脑脊液化验IgG合成率1. 9mg/dl(-9. 9~3. 3),BB 12. 0(<7. 4×10-3),Index 1. 96(<0. 7),寡克隆区带血清脑脊液阳性。髓鞘碱性蛋白(血清) 5. 5(<20ng/ml),髓鞘碱性蛋白(脑脊液)<1. 0(<5. 0ng/ml)。
6.心电图 见窦性心动过速,ST-T改变。
7.腹部超声 见脂肪肝。
8.胸部CT(2009年9月28日)胸腺区未见异常密度影。气管分叉上方局部腔内小结节,陈旧改变可能性大。
9.全身骨扫描 未见明显骨异常征象。
10.双下肢胫部MRI扫描 未见异常。
11.新斯的明试验(2009年4月)“阳性”,具体不详。我院复查,肌肉注射甲硫酸新斯的明注射液1. 5mg+硫酸阿托品注射液1. 0mg后,症状改善不明显。
12.针极肌电图(2009年8月27日、9月4日、9月24日)广泛神经源性损害; (2009 年9月28日,解放军第309医院)考虑神经源性受损伴神经性受损。
13.感觉神经传导:左侧腓肠神经传导速度减慢;运动神经传导:双侧胫神经复合肌肉动作电位(CMAP)波幅减低,双侧腓总神经CMAP波幅显著减低,右侧腓总神经远端CMAP未引出。
14.重复神经刺激 右侧副神经、腋神经低频刺激时波幅出现递减现象(低频递减超过15%),高频刺激时波幅未见明显递增递减现象。
【诊治经过】
2009年11月12日给予糖皮质激素冲击(注射用甲泼尼龙琥珀酸钠1. 0g× 3天,0. 5g×3天,0. 25g×3天,120mg×3天,60mg×3天,改口服醋酸泼尼松片40mg渐减量)、营养周围神经(维生素B1,甲钴胺)、降血脂(阿托伐他汀钙片20mg,1次/晚),益气扶正(参芪扶正注射液)、改善微循环(前列地尔注射液、注射用盐酸川芎嗪)等治疗。
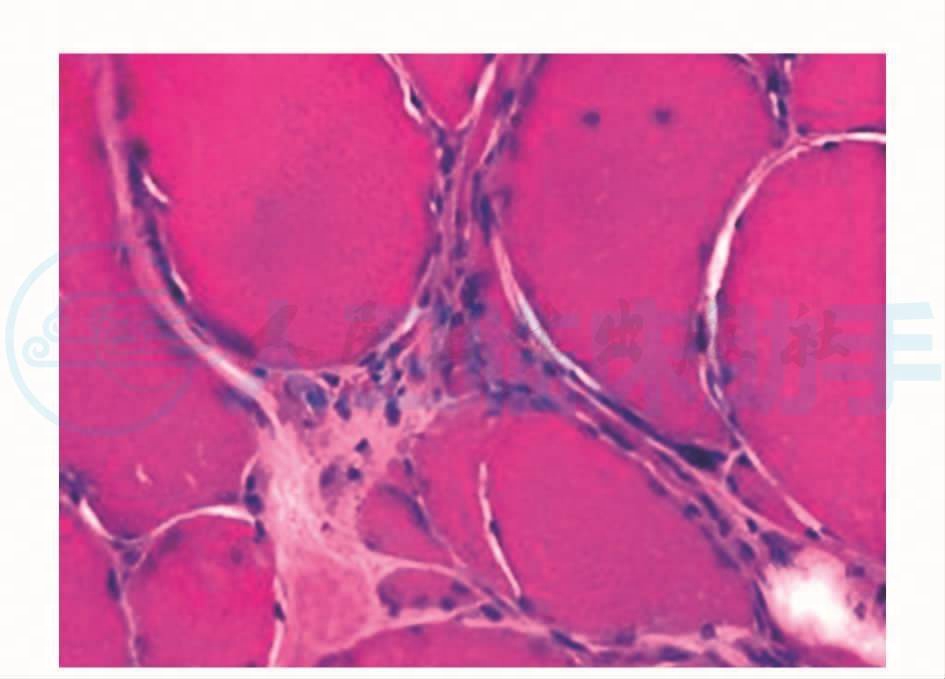
图1 肌纤维胞浆膜下镶边空泡,肌间质内见炎性细胞浸润,HE×200
【病理结果】
取材部位:左腓肠肌+腓肠神经
1.左腓肠肌组织学和组织化学检查结果
组织学检查(HE、MGT、PAS和ORO染色)
(1)HE:
肌肉横切面显示数个边界清楚的肌纤维束,间质内脂肪结缔组织无明显增生。小血管壁结构正常,在部分区域的肌间质内可见到炎性细胞浸润(图1),但未见到炎性细胞浸润到非坏死肌纤维中。可见到成束分布和呈网状分布的小角状萎缩肌纤维,以及小圆状萎缩肌纤维,部分肌纤维肥大变圆。部分肌纤维内可见到镶边空泡(RV) (图2)。可见到1个坏死的肌纤维。个别肌纤维的胞浆出现嗜碱性改变(图3)。部分肌纤维出现核内移现象。未见到环状和旋涡状肌纤维。
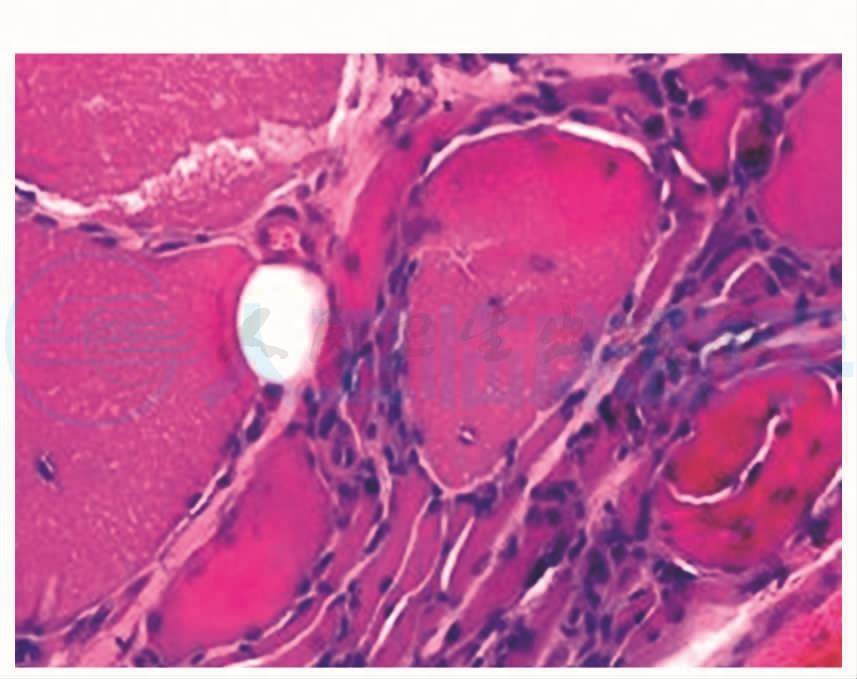
图2 见成束分布和呈网状分布小角状萎缩肌纤维,部分肌纤维肥大变圆。可见镶边空泡,HE×200
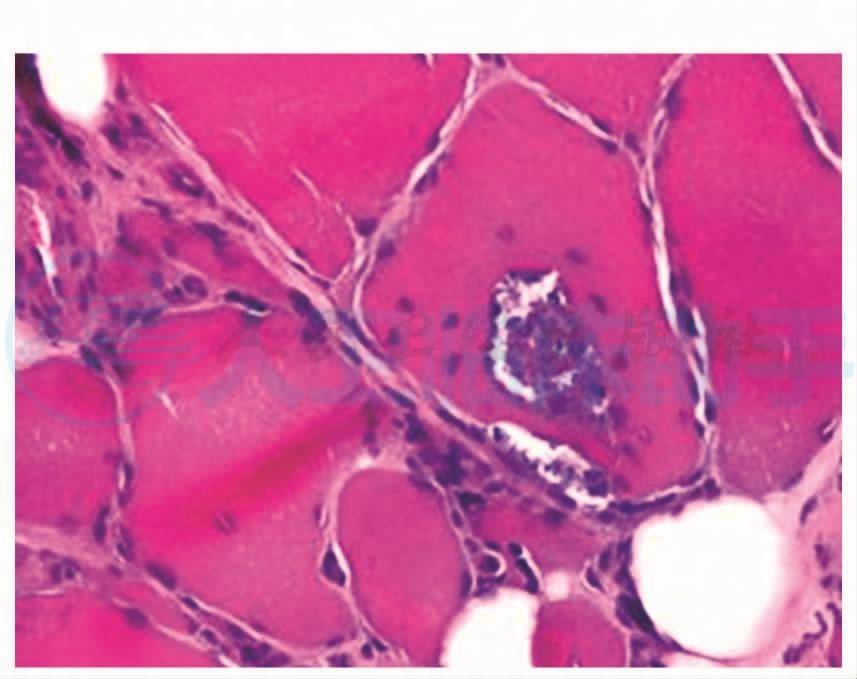
图3 见肌间质内炎性细胞浸润,个别肌纤维胞浆出现嗜碱改变,部分肌纤维可见镶边空泡,HE×200
(2)MGT:
未见到典型或不典型的RRF。
(3)PAS:
个别肌纤维内糖原呈条索状沉积。
(4)ORO:
个别肌纤维内脂肪滴轻度增多。
酶组织化学检查(肌丝ATP酶酸碱系列,NADH-TR、SDH和NSE染色)
(1)ATP酶酸性反应:
显示Ⅱ型肌纤维病理性占优,仅见到个别散在分布的Ⅰ型肌纤维,萎缩和肥大肌纤维累及两型。ATP酶碱性反应:证实肌纤维的上述分布特点。
(2)NADH-TR:
个别萎缩的肌纤维出现轴空样改变,极度萎缩的肌纤维深染。
(3)SDH:
未见到深染的肌纤维以及深染的血管。
(4)NSE:
可见到萎缩的肌纤维深染,肌间小血管未见深染。
骨骼肌呈肌病样病理改变伴随神经源性改变
骨骼肌主要病理改变是肌纤维出现萎缩、再生、肥大、坏死、炎细胞浸润以及肌纤维内镶边空泡,符合空泡性肌肉病的病理改变特点。出现成束分布和呈网状分布的小角状萎缩肌纤维,以及肌纤维出现群组化现象,提示存在神经源性骨骼肌损害的病理改变特点,结合患者的临床特点不除外包涵体肌炎或包涵体肌病的可能性。
2.左腓肠神经半薄切片检查结果
见10个横切的神经束,神经外衣的结缔组织中可见少量脂肪细胞和数个小血管。血管壁内皮细胞及平滑肌细胞明显肿胀。小血管周围无炎细胞浸润。神经束衣无明显增厚,神经束内有髓神经纤维数量轻度减少。可见较多薄髓鞘的有髓神经纤维,以及有髓神经纤维分裂现象。可见有髓神经纤维成簇排列现象。没有发现有髓神经纤维变性形成的髓球样结构。也没有见到有髓神经纤维周围出现洋葱球样结构。无髓神经纤维无明显减少。未见神经内衣结缔组织增生以及炎细胞浸润。神经束衣内出现水肿改变。毛细血管基底膜不增厚。病理诊断:混合性周围神经病理改变(左腓肠神经)。评判:周围神经存在明显水肿改变,可能与人工假象有关。周围神经可见薄髓鞘神经纤维以及有髓神经纤维再生现象,提示混合性周围神经病理改变。上述病变多见于获得性周围神经病。









