


 收藏
收藏 已收藏
已收藏【主诉】
进行性四肢麻木、疼痛、无力、肌肉萎缩2年半,加重半年。
【现病史】
患者男性,45岁,天津人,工人。患者于入院前两年半(1983年底)开始自觉双小腿紧缩感,双足底针刺样疼痛,受凉后明显,伴有稀便,2~3次/天,无发热、腹痛不适。当地医院给予肌注维生素B1、B12治疗10天后症状无改善,感觉异常逐渐加重并累及上肢,感四肢无力,尤以下肢为重。于1984年底再次就诊当地医院,入院后行腰椎穿刺术,提示脑脊液蛋白0. 92g/L,余未见异常;肌电图示神经源性损害;考虑“周围神经病变”,给予地塞米松、B族维生素及中药等治疗无效。症状进行性加重,四肢肌肉逐渐萎缩,并出现水样便,最多6~7次/日,有时大便失禁,并相继出现颜面及双踝部水肿,排尿费力,走路不稳及站立时头晕。入院前半年,坐位时亦感头晕,先后晕厥3次而被迫卧床。
【过去史及个人史】
无特殊。
【家族史】
祖父、父亲均为独生子。其祖父因患腿痈病3年,45岁病故。父亲因四肢麻木、疼痛、无力、肌肉萎缩、腹泻、立位时头晕、多次晕厥等3年,于47岁因心衰突然死亡。两人曾用秋水仙碱治疗症状有缓解。患者兄弟四人,弟弟及子女无任何自觉症状,全部进行了内科及神经系统检查,包括血常规、肝肾功能、血免疫球蛋白、胸部X线透视、心电图、超声心动图、腹部B超,四肢的感觉、运动神经传导检查。除41岁的大弟弟脾厚4. 4cm,实质内可见散在点状强回声,38岁的二弟弟脾厚4. 0cm,实质内可见点状强回声外,余无阳性所见(附家系图1)。
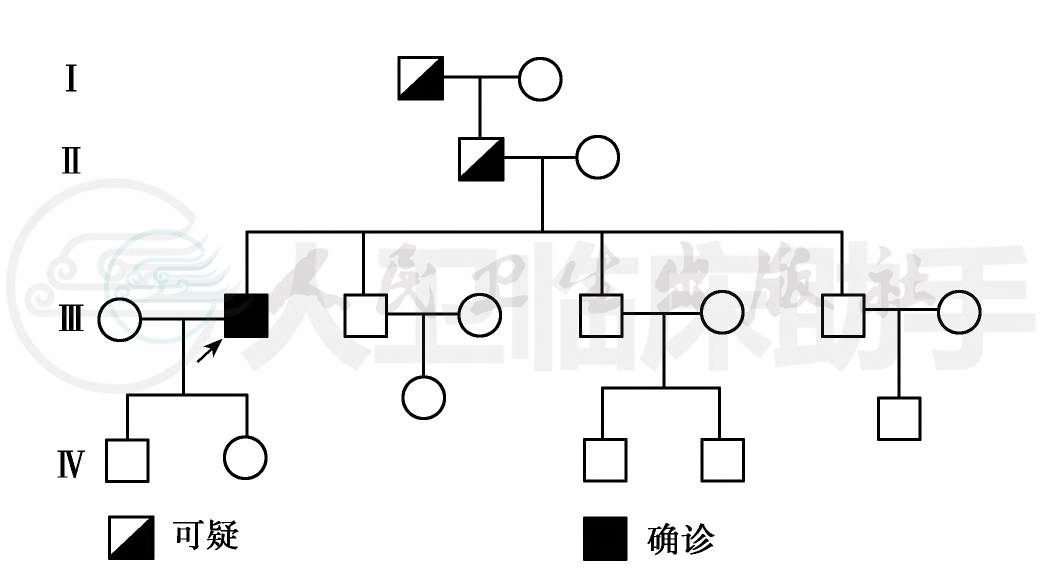
图1 患者家系图
【查体】
卧位血压89/60mmHg、坐位血压40/30mmHg。慢性消耗病容,棕褐色皮肤以面部为著,颜面及双踝部水肿。舌体积正常,心率60次/分,偶有早搏。神经系统查体:意识清楚,语言流利,脑神经检查未见异常。四肢肌肉萎缩,双上肢肌力4级,双下肢肌力2~3级,远端重。四肢肌张力低。双膝以下痛觉减弱,位置觉及振动觉消失。腱反射消失,双侧病理征阴性。
【辅助检查】
1.尿便常规正常。血常规:红细胞计数4.3×1012/L、白细胞计数3.5×109/L、血红蛋白112.9g/L、血小板计数162×109/L;尿、便常规正常;尿本周氏蛋白阴性。
2.血生化、血糖、肝肾功能正常,A/G为2.9/2.3,血蛋白电泳γ 183%,血IgG 130U/ml(正常140U/ml±404U/ml)、IgA 58U/ml(正常120U/ml±40U/ml)、IgM 42U/ml(正常160U/ ml+70U/ml)、IgD 21U/ml(正常10U/ml~100U/ml),ERFC 40%(正常50%~70%),LTT 47%(正常50%~80%),T4 25%、T8 23%(正常T4/T8为2/1)。血康华反应、类风湿因子、抗核抗体及LE细胞均阴性。
3.脑脊液检查 Pandy试验(+),蛋白1. 5g/L,余指标正常。
4.骨髓片 正常。
5.心电图 心电图左前半阻滞,异常Q波,电轴左偏。心机械图示不正常心功能。
6. X线检查 胸片示心脏呈普大型扩大,头颅、腹部及骨盆X线检查无异常。
7.超声心动图 示全心肌肥厚、心包积液、前心包暗区0. 6cm,后心包暗区1.6cm;
8.腹部B型超声检查 无异常。
9.电生理检查 脑电图正常;肌电图神经源性损害,感觉运动传导速度减慢。
10.头颅CT扫描 未见异常。
【诊治经过】
入院后行右腓肠肌和右腓神经活检,病理证实为淀粉样多发神经病,结合阳性家族史确诊为家族性淀粉样多发神经病。10月6日开始用秋水仙碱0. 5mg 3次/日,同时配用多种维生素。颜面及双踝部水肿消退。2个月后复查超声心动图,心包积液基本消失,胸片心脏明显缩小。5个月后复查脑脊液蛋白降至1. 06g/L,余自觉症状及体征无明显变化。住院238天自动出院回家。
【病理结果】
取材部位:右腓肠肌和右腓肠神经。
右腓肠肌活检:石腊切片HE染色,肌纤维明显弥漫性萎缩;冰冻切片酶组化染色: ATP酶在pH 9. 6、4. 6、4. 2环境中,因肌肉明显萎缩,不能分辨其纤维类型。甲苯氨兰、结晶紫及刚果红染色:于肌纤维间质和小血管周围有淀粉样物沉积。电镜:萎缩的肌纤维内,间质中及小血管周围均可见到纤细的、排列紊乱的淀粉样物质。肌节紊乱,个别肌细胞基底膜增厚。
右腓肠神经活检:刚果红、甲苯氨兰及结晶紫染色,可见神经束内外均有淀粉样物沉积,同时可见到施万细胞增生,较多轴索消失,束间小血管周围可见小圆细胞浸润。
FAP无特效疗法,迄今尚无特异性消退淀粉样物质沉积灶的方法。目前以对症支持疗法为主,临床试用的药物及肝移植主要是针对TTR基因突变造成的本病。秋水仙碱在阻止淀粉样蛋白的合成和促排方面可能有一定的作用。本例用秋水仙碱治疗有效,未发现明显副作用。也有人用二甲基亚砜(DMSO)治疗,DMSO是工业溶剂,首先被Lsobe和Osserman用来治疗淀粉样变性病,能使乳酪诱发的实验小鼠的淀粉样蛋白沉积部分和全部消失。双氯芬酸和氟比洛芬能抑制转甲蛋白的裂解,稳定四聚体,防止转甲蛋白淀粉样纤维在体内形成和沉积。肝移植在国外是从1990年开始运用于临床的,用于治疗由TTR变异导致的FAP,肝移植切除突变蛋白的合成部位对遗传性淀粉样变性进行治疗已取得很好的疗效。目前肝移植已经是公认的治疗FAP的一种方法。Okamoto等对瑞典108例进行肝移植和33例没有进行移植的研究分析发现,与对照组比较,肝移植组患者的生存率显著提高,特别是对于早发型患者(<50岁)生存率有一个特别显著的差异(P<0.001)。相反,而对于晚发型患者(≥50岁)两者没有显著差异。与迟发型男性相比,迟发型女性肝移植术后生存率有显著提高(P=0.02)。但是对于患病7年以下和7年以上的两组中发现肝移植对生存率没有显著差别,迟发型男性患者肝移植后生存率也没有改善。另外通过核酶对TTRmRNA的降解来减少TTR的产生,是一种很有潜力的基因治疗方法。
分子诊断的应用为FAP的发病机制研究以及疾病的有效诊断、早期干预提供了极大的帮助。随着分子生物学技术的不断发展以及新型基因治疗、纳米靶向治疗等新技术的应用,必将为FAP提供有效的诊断、治疗手段。









