


 收藏
收藏 已收藏
已收藏男,17岁,汉族人,学生。
主诉
间歇性呕吐6年,肢体无力2年,缓解后复发6个月
患者前两次肌肉活检均为石蜡切片,石蜡切片因需要经过二甲苯固定、加热等的过程,肌肉的形态、结构和酶的活性会受到一定破坏,所以肌肉活检石蜡切片早在20世纪60年代已经淘汰,肌肉活检应行冰冻切片检查。另外,肌肉活检组织病理学固然重要,但将诊断止步于病理诊断现在看来是远远不够的,应探究其生化和分子病理学基础。
病史
患者6年前无明显诱因出现反复恶心、呕吐,呕吐物为胃内容物,呕吐物内未见隔夜宿食,经补液等对症处理后约1~2天好转,此后于每年秋天均有类似发作,发作时频繁呕吐,每日4~5次,如未经治疗,持续1周左右可自行缓解,间歇期约为1个月,曾在当地医院行钡餐、胃镜等检查,诊为“胃炎、食管裂孔疝”,予以抗酸、增强胃动力类药物治疗无效。2年前呕吐频繁,间歇期缩短为15天,非喷射性,并出现四肢乏力,双肩疼痛,易疲劳,病情进行性加重。1年半前曾在北京某医院住院,查CK 2286U/L,行腓肠肌活检石蜡切片检查,病理报告符合“肌源性损害”,遂按“多发性肌炎”,给予泼尼松20mg,每天3次,环磷酰胺100mg,每天1次,治疗1个月后肌无力症状有所缓解,血CK降至760U/L。继续上述治疗1个月后逐渐减量激素,其间症状时好时坏。6个月前泼尼松减至15mg/d,并停用环磷酰胺,症状再次加重,四肢、颈项部无力,抬头及咀嚼费力,下蹲后起立困难,上楼困难,复查CK又升至 2424U/L,遂再次回某医院住院治疗,并第二次行腓肠肌活检,病理报告为“部分肌纤维变性,个别肌纤维坏死,肌膜核增生、内移,间质及小血管未见炎性细胞,符合肌源性损害”,继续给予泼尼松15mg/d,肌力无明显改善,复查CK 1443U/L,出院时泼尼松减至10mg/d,1个月后减至5mg/d。患者肌无力症状仍进行性加重,于停药2个月后来我院就诊。当时神经系统查体:患者神志清,言语及定向力正常,肥胖体形,Cushing面容,腹部、肩部和大腿内侧皮肤可见大片皮纹,查体配合。脑神经外观(-),颈伸肌、咬肌无力,四肢近端肌力4级,远端肌力5级,不能跳跃。无明显肌萎缩和肌束颤动。四肢腱反射减弱(+/-),无锥体束征,无感觉障碍,肌肉无压痛。脑膜刺激征(-)。辅助检查:血常规大致正常。肌酶谱:CK 1532U/L(25~200U/L),AST 172U/L(1~40U/L),LDH 895U/L(120~230U/L)。空腹血糖 3.25mmol/ L(3.9~6.0mmol/L),血酮体(-)。肌电图未见异常。风湿系列无明显异常。甲状腺功能正常。胸片未见异常。心电图正常。上消化道钡餐未见器质性病变。
既往史
无肝炎、结核病史,无食物和药物过敏史,除不喜食肥肉外无其他偏食习惯。
家族史和个人史
患者为独生子,父母均健康。家族中无类似患者可查。
线粒体神经胃肠脑肌病(MNGIE)是线粒体脑肌病的一个亚型,属常染色体隐性遗传。多于青少年发病,主要临床症状包括胃肠道动力障碍、恶液质、眼外肌麻痹、周围神经病及白质脑病,肌肉病理可见RRF和COX缺失纤维。本病由于胃肠道动力障碍非常明显而常被误诊。
第一个诊断
线粒体脑肌病可能性大。结合间歇性呕吐及进行性四肢无力,而且两次活检均显示肌源性损害,激素治疗似乎有短期效果,所以线粒体脑肌病不能除外,其中以线粒体神经胃肠脑肌病(MNGIE)可能性最大。但两次活检未见RRF,这一点不是很支持。但并非所有的线粒体脑肌病都必须有RRF,有一部分患者仅有COX缺失或脂质的沉积。门诊的处理是给予辅酶Q10及肌苷口服。考虑到前两次活检均为石蜡切片,建议患者再次行肌活检术。
第二个诊断
脂质沉积性肌病需除外。患者系少年期起病,病程迁延,肌无力症状以颈伸肌、咀嚼肌和肢体近端为主。肌无力是神经肌肉病常见的症状,不同类型的肌肉病,其肌无力的分布也有一些规律可循(表1),颈伸肌、咀嚼肌受累往往多见于肌炎、脂质沉积性肌病和甲减性肌病,患者的甲功正常从而后者可以排除,而多发性肌炎的诊断在18岁之前的患者基本不予考虑。
表1 常见肌肉病肌无力的分布特点

在征得患者及家属同意后在我院再次行肌活检术。活检部位为左侧肱二头肌。标本行连续冰冻切片。HE染色见肌纤维大小不等,大量肌纤维内可见细小圆形空泡,有的相互融合成不规则大泡,未见坏死和再生纤维,肌内膜无增生,未见炎症细胞浸润。MGT染色:未见典型的不整红边纤维(RRF),未见镶边空泡(rimmed vacuole),无胞浆体。NADH染色:肌原纤维间网格样结构排列紊乱,Ⅰ型纤维明显:可见粗大深染颗粒,无同型纤维群组化现象。SDH染色:酶活性明显减低,Ⅰ型纤维内可见深染的颗粒,未见SSV。COX染色未见酶活性缺失。PAS染色:糖原轻度增加。ORO染色:大量肌纤维内脂质明显增多,以Ⅰ型纤维尤为显著。病理诊断:脂质沉积性肌病(病理见图1)。
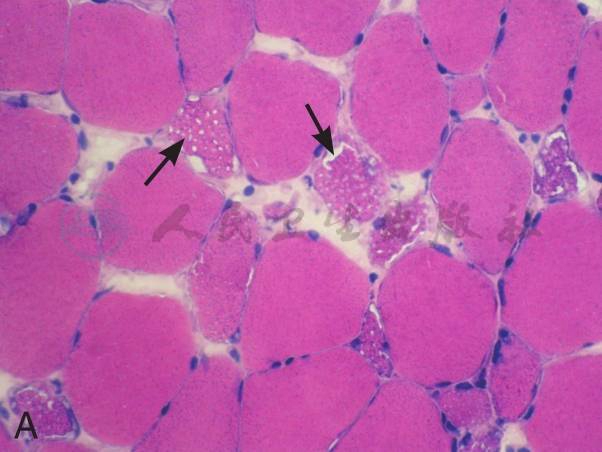
A:HE染色示肌纤维大小不等,大量肌纤维内可见细小圆形空泡,有的融合成不规则大泡(如箭头所示,×400)
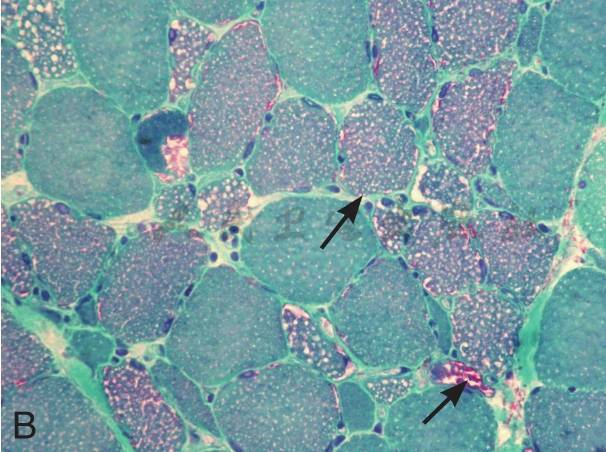
B:MGT染色:可见大量红染纤维(如箭头所示),但未见典型的RRF
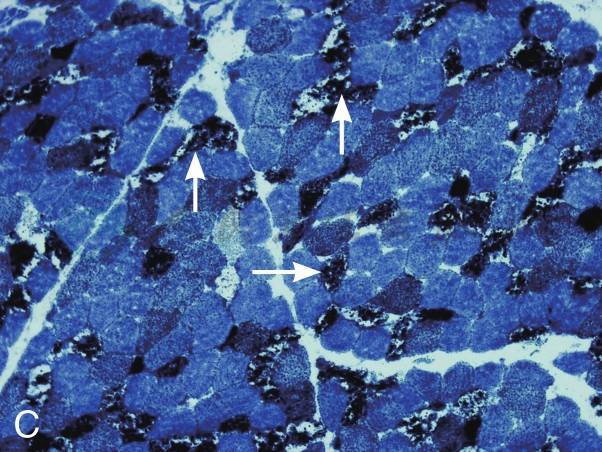
C:NADH染色:肌原纤维间网格样结构排列紊乱,大量纤维深染(如箭头所示,×100)

D:SDH染色示酶活性明显减低,Ⅰ型纤维内可见深染的颗粒(如箭头所示,×400)
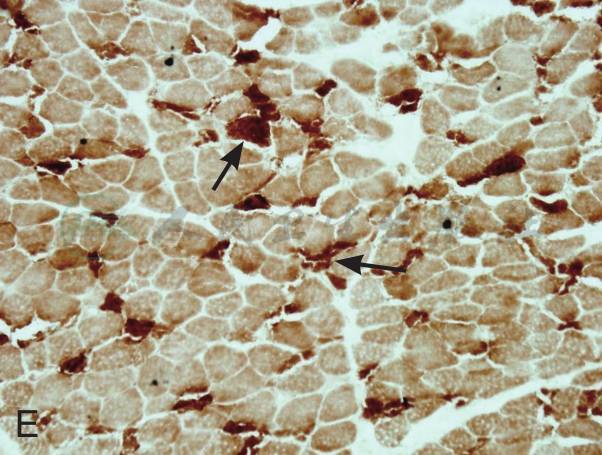
E:COX染色未见酶活性缺失,大量肌纤维深染(如箭头所示,×100)
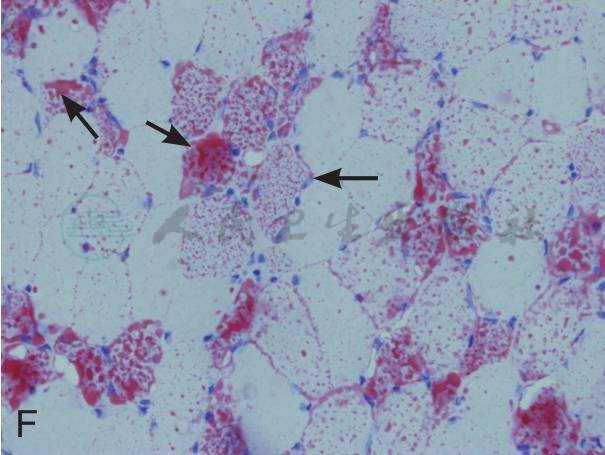
F:ORO染色大量肌纤维内脂质明显增多(如箭头所示,×400)
图1 患者肱二头肌活体组织检查病理结果
以戊二酸尿症Ⅱ型为代表的有机酸尿症是遗传代谢病的一个重要组成部分,因为长期的有机酸血症会造成中枢神经系统损伤,所以又称脑有机酸血症。
为筛查与脂质代谢有关的遗传代谢病,搜集患者的空腹尿和血标本送检尿有机酸和血脂酰肉碱分析。GC-MS尿有机酸分析结果提示:①枫糖尿症的二氢脂酰脱氢酶(E3)缺乏②2-羟基戊二酸尿症③戊二酸尿症Ⅱ型(glutaric aciduria type Ⅱ)(图2)。血滤纸片脂酰肉碱检测结果(日本MILS实验室):血中C8、C10、C12脂酰肉碱浓度分别为1.04、1.43和0.63nmol/ml,较正常对照明显增加,血中游离肉碱浓度为30.5mmol/L,较正常对照降低,生化诊断:戊二酸尿症Ⅱ型(GAⅡ),又称多种酰基CoA脱氢缺陷(MADD)。
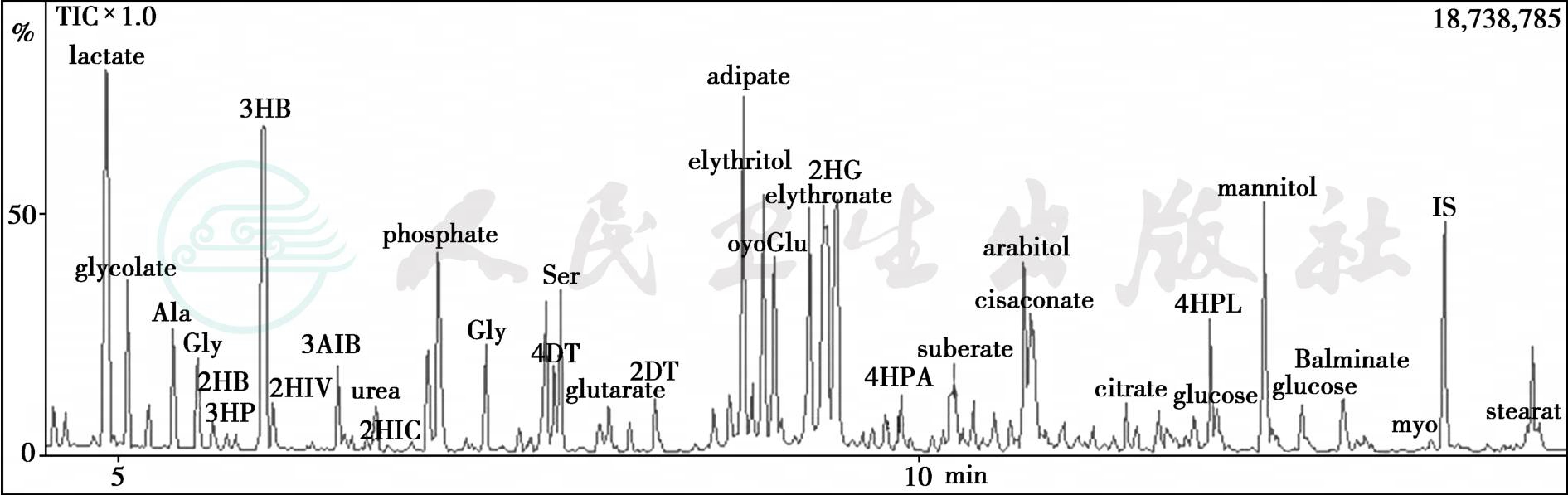
图2 GC-MS尿有机酸分析结果
为探究其分子病理学基础,在征得家属及患者同意后行皮肤活检,患者的皮肤成纤维细胞培养电子转运黄素蛋白(ETF)或电子转运黄素蛋白脱氢酶(ETF-DH)基因分析结果:ETF基因未见突变;ETF-DH基因有两个位点的突变:A1227C(L409F,外显子10),即亮氨酸突变为苯丙氨酸;G1399C(G467R,外显子11),即甘氨酸突变为精氨酸,故该患者的分子病理诊断为ETF-DH基因突变。
单纯给予补充维生素B2 50mg,每天3次,并嘱多食用瘦肉、高碳水化合物饮食及新鲜绿叶蔬菜,1周后肌无力症状明显缓解,1月后肌力已基本接近正常,1年后停用维生素B2,仅饮食调节,随访4年未再复发。后来又对其父母进行了尿有机酸的检测,其结果母亲是正常的,而其父亦存在轻度的戊二酸尿症,故嘱其注意饮食调整。









