


 收藏
收藏 已收藏
已收藏瓦登伯格综合征(Waardenburg syndrome,WS)是一组与全身或眼部色素变化有关的综合征,包括皮肤或头发的脱色素改变,眼部蓝色外观和虹膜异色,听力下降,其他WS亚型的眼部和全身异常还包括内眦外移,四肢肌肉骨骼发育异常,先天性巨结肠,神经系统异常。WS是具有遗传异质性的常染色体显性遗传疾病。
WS患病率约为1/40000,是一种罕见的遗传性疾病。按照临床特征和遗传异质性,WS分为四种类型,WS1~WS4的基因变异类型分别为paired box 3 (PAX3),melanogenesis-associated transcription factor (MITF),endothelin 3 (EDN3),endothelin receptor type B (EDNRB),snail-family transcriptional repressor 2 (SNAI2),SRY-box 10 (SOX10)。 WS1和WS2是WS中比较常见的类型,WS4是一种比较罕见的亚型。WS4也被称为Waardenburg-Shah综合征,临床特征为听力下降或丧失、全身色素脱失表现,合并先天性巨结肠。WS4型又分为WS4 A~C三型,分别由EDNRB、EDN3和SOX10突变引起。EDNRB和EDN3突变分别以常染色体隐性(autosomal recessive,AR)和常染色体显性(autosomal dominance,AD)方式遗传。SOX10突变以常染色体显性方式遗传。SOX10是一个关键性转录因子,在神经嵴衍生细胞迁移和分化过程中,与MITF、酪氨酸酶、髓鞘蛋白及连接蛋白β1和EDNRB的靶向活动有关。
本病例介绍1例瓦登伯格综合征Ⅳ型合并开角型青光眼患者的临床特征和基因变异类型。患者基因检测到EDNRB基因的杂合缺失突变,这种突变分别来自患者父亲和母亲,其父母未发病。
患者男性,43岁,自幼双眼球“发蓝”,视野渐进性缩小。全身检查头发及身体皮肤没有明显脱色素改变。听力检测、头颅MRI检查及全身体检未见明显异常。双眼结膜下滤过泡限局性隆起,双眼虹膜异色,双眼底弥漫性视网膜色素脱失呈晚霞样外观,视盘盘沿弥漫性缩窄。前节OCT(AS-OCT)在虹膜异色部位虹膜前界膜呈低反光表现。患者和其父母发现EDNRB基因2个杂合突变位点。临床诊断:瓦登伯格综合征Ⅳ型,合并开角型青光眼。治疗为控制眼压双眼分别行滤过性手术,术后眼压控制在11~14mmHg之间,视野及视神经损害未再发生进展。
瓦登伯格综合征;开角型青光眼;EDNRB基因;晚霞样眼底
男性患者,出生后其母亲发现双眼睛发蓝,17岁时发现双眼的眼压升高至26~30mmHg之间,药物控制眼压不佳,视野渐进性缩窄,手术治疗,右眼行非穿透性小梁手术,眼压控制半年后上升,复查时在房角镜下右眼非穿透手术内口激光切开,术后眼压控制。左眼行小梁切除术,术后随诊,眼压控制,不需要应用降眼压药物的情况下,眼压在12~16mmHg之间。双眼的滤过泡形态及功能良好。
视力:右眼0.05,矫正(-9.0D)0.4,左眼手动/眼前,矫正不提高。双眼球水平震颤,左眼外斜视,双眼无内眦外移。
裂隙灯检查:双眼滤过泡限局性隆起,角膜清,前房中深,周边前房深度大于1/2CT,右眼部分虹膜异色,3:30到8:30,以及10:30到12:00之间的虹膜色泽正常;左眼大部分虹膜异色,仅留1:00到2:00之间1个时钟位虹膜色泽正常。晶状体前囊膜表面有色素沉着(图1)。
眼底像检查:视网膜色素脱失,双眼底呈晚霞样外观,视盘盘沿弥漫性缩窄,视网膜神经纤维层弥漫性丢失,晚期青光眼视神经损害(图2)。
房角镜检查:小梁网明显色素沉着,色素Ⅳ级,虹膜与小梁网夹角为45°。在房角镜下可见虹膜周边粗大的血管(图3,图4)。
前节OCT(AS-OCT)可以看到双眼虹膜的结构变化。右眼虹膜颞侧包含有正常虹膜和异色虹膜组织,对比发现正常虹膜的前界膜反光较强,遮挡前界膜下方的虹膜组织结构的反光,虹膜后色素上皮层几乎不可见;而在右眼异色虹膜部位,由于虹膜前界膜组织黑色素缺失,前界膜组织变薄,光反射信号减弱,前界膜下方组织的反光增强,后色素上皮层光反射信号增强,可清晰显示虹膜后色素上皮层结构。右眼虹膜鼻侧和颞侧均有异色虹膜和正常虹膜,扫描可见异色部位前界膜的低反光和清晰的下方虹膜结构反光及正常虹膜的前界膜高反光和较弱的下方结构反光的翻转对比(图5,图6)。
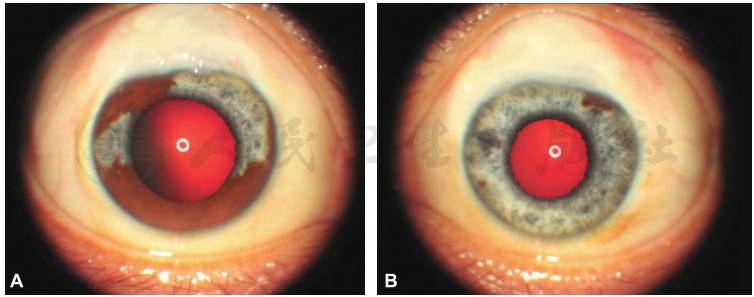
图1 外眼像检查
外眼像检查显示双眼滤过泡限局性隆起(A,B),右眼部分虹膜异色,3:30到8:30,以及10:30到12:00之间的虹膜色泽正常(A);左眼大部分虹膜异色,仅留1:00到2:00之间1个时钟位虹膜色泽正常(B);双眼外眼像(C)显示右眼不完全性虹膜异色,左眼近完全性虹膜异色。

图2 眼底像检查及后节OCT
A、B.视盘周围2~3D范围视网膜色泽正常,由于大部分视网膜色素脱失,双眼底呈晚霞样外观,视盘盘沿弥漫性缩窄,视网膜神经纤维层弥漫性丢失,晚期青光眼视神经损害;C、D.后节OCT显示,脉络膜和视网膜组织变薄。
引自:主编:.同仁眼科疑难病例精析:同仁眼科临床病例讨论会1.第1版.ISBN:978-7-117-33030-5
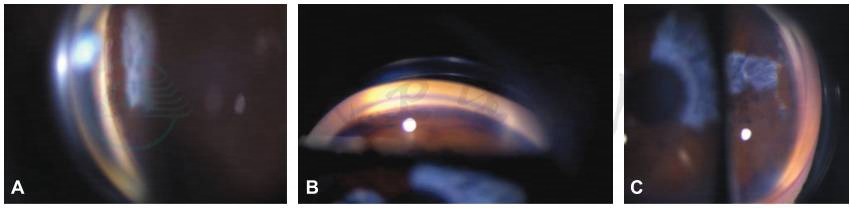

图3 右眼外眼像及房角镜检查
4个象限房角开放,小梁网色素沉着,虹膜与小梁网夹角为45°;A.鼻侧房角;B.下方房角;C.颞侧房角;D.上方房角,可见滤过手术的内口;E.右眼外眼像。

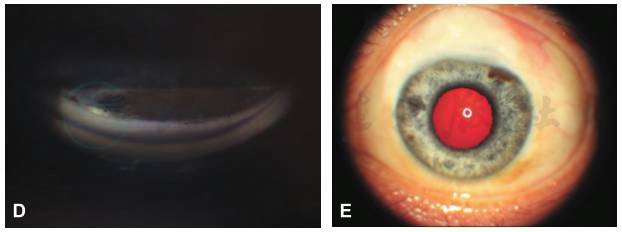
图19-4 左眼外眼像及房角镜检查
4个象限房角开放,小梁网色素沉着,虹膜与小梁网夹角为45°;下方可见虹膜周边粗大的血管;A.颞侧房角;B.下方房角;C.鼻侧房角;D.上方房角,可见滤过手术的内口;E.左眼外眼像。
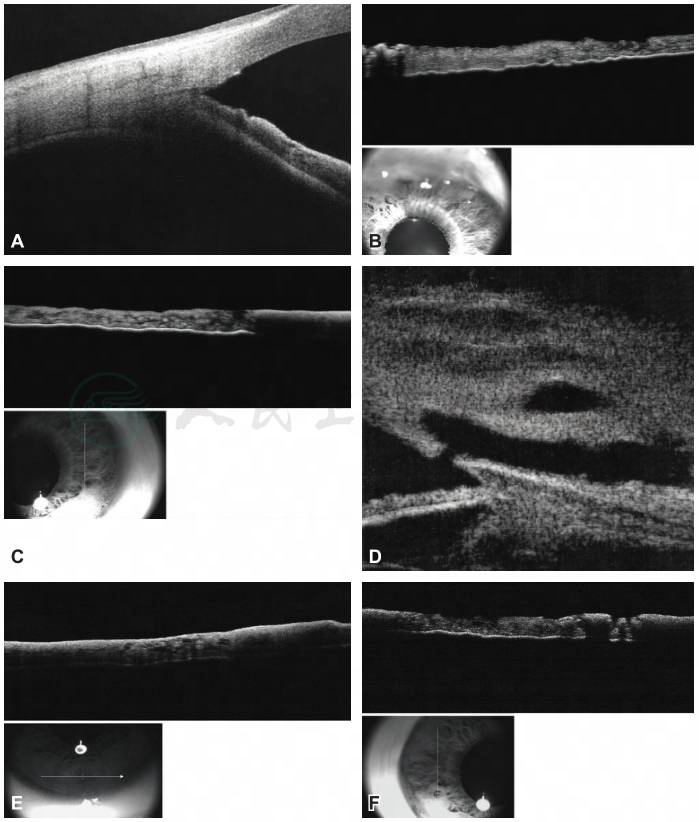
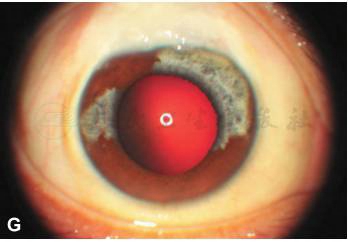
图5 前节OCT(AS-OCT)及UBM检查显示右眼虹膜及滤过泡结构变化
A.房角开放;B.上方部分虹膜组织,有色素的虹膜前界膜萎缩,由于虹膜前界膜组织黑色素缺失,前界膜组织变薄,光反射信号减弱,所以前界膜下方组织的反光增强,可清晰显示虹膜后色素上皮层结构;C、F.右眼鼻侧虹膜(C)和颞侧虹膜(F),正常虹膜和异色虹膜组织,对比发现正常虹膜的前界膜反光较强,遮挡前界膜下方的虹膜组织结构的反光,虹膜后色素上皮层几乎不可见;而在异色虹膜部位,由于虹膜前界膜组织黑色素缺失,前界膜组织变薄,反光减弱,所以前界膜下方组织的反光增强,可清晰显示虹膜后色素上皮层结构;异色虹膜和正常虹膜间组织延续,扫描可见异色部位前界膜的低反光和清晰的下方虹膜结构反光及正常虹膜的前界膜高反光和较弱的下方结构反光的翻转对比;D.右眼滤过泡通道形态正常;E.下方虹膜正常,正常虹膜的前界膜反光较强,遮挡前界膜下方的虹膜组织结构的反光,虹膜后色素上皮层几乎不可见;G.右眼外眼像,部分虹膜正常,3:30到8:30,以及10:30到12:00之间的虹膜色泽正常。

图6 前节OCT(AS-OCT)显示左眼虹膜结构变化
左眼大部分虹膜异色,仅留1:00到2:00之间1个时钟位虹膜色泽正常;由于左眼大部分虹膜的前界膜组织黑色素缺失,前界膜组织变薄,光反射信号减弱,所以前界膜下方组织的反光增强,可清晰显示虹膜后色素上皮层结构,后色素上皮层光反射信号增强;
引自:主编:.同仁眼科疑难病例精析:同仁眼科临床病例讨论会1.第1版.ISBN:978-7-117-33030-5
后节OCT显示,双眼脉络膜和视网膜组织变薄,视神经乳头周围神经纤维层分析显示双眼视网膜神经纤维层(retinal nerve fiber layer,RNFL)厚度变薄,右眼平均RNFL厚度47μm,左眼49μm (图7A)。Humphery视野检测显示右眼MD值为-13.52dB(图7B),左眼MD值-27.87dB(图7C)。


图7 双眼后节OCT及视野检查
后节OCT显示,双眼脉络膜和视网膜组织变薄,视神经乳头周围神经纤维层分析显示双眼视网膜神经纤维层(RNFL)厚度变薄(A);双眼视野检测显示右眼视野中度损害(B),左眼视野重度损害(C)。
患者听力检测正常。颅脑CT及MRI检测均未见明显异常。
患者父母及儿子均接受全身及眼部检查,父母除白内障外无其他眼部异常。全身检查除患高血压外未见其他明显异常。患者的儿子也接受全身及眼科检查,除双眼屈光不正(近视)外其他均未见明显异常。
患者及家属接受基因检测前均签署了知情同意书。患者、其父母及患者的儿子均采集EDTA抗凝血液标本。应用Panel测序及外显子测序方法,分析患者及其家属的基因变异类型,采用常规CTAB法提取样品基因组DNA,并对样品DNA进行质量检测。捕获建库。目标基因区域捕获测序。使用引物库进行引物设计,并使用UCSC的PCR功能,查看引物的退火温度及序列的复杂性。对于引物库中没有的染色体位点,通过UCSC进行序列查找,并用Primer3.0进行引物设计,引物合成。分别进行传统PCR(Sanger法)测序和外显子组测序筛选变异。经PCR扩增,PCR产物分析,上机测序操作,生物信息数据分析,高通量测序得到的原始图像数据文件经碱基识别分析转化为原始测序序列。用Fastq-Mcf将短序列、引物自连、去掉低质量序列得到预处理后数据,比对到参考基因组(UCSC GRCh37/hg19),得到含有比对信息的SAM/BAM格式文件,进行变异检测。去除接头序列和低质量碱基的测序数据用Burrows-Wheeler Aligner(BWA) 与人类基因组比对;使用GATK(genome analysis toolkit)软件包对质量值进行校准后重新匹配到参考序列上,找出序列中的SNV(single nucleotide variation)及 InDel(insertion-deletion);利用CCDS、人基因组数据库(HG19)、dbSNP(v138)等信息对变异进行注释,确定突变位点在参考序列中的位置、效应等信息;SIFT预测SNP影响蛋白功能。
患者及其亲属的基因变异类型结合患者的临床表型,确定患者的临床诊断为瓦登伯格综合征(Waardenburg syndrome)合并开角型青光眼。在患者及其父母基因型中发现有EDNRB基因2个变异位点,第1个变异是外显子exon 7 c.1111G>A,导致氨基酸在密码子371位点从甘氨酸变为丝氨酸。这个变异类型在1000 Genome,ESP6500,ExAC_ALL和 ExAC_EAS人群基因组数据库中并未发现,为证实c.1111G>A (p.G371S)改变是EDNRB基因变异类型,并与患者临床表型直接相关,患者父母同样也做了Sanger测序分型,Sanger证实基因突变来源于其母亲(图8);第2个变异位于c.553G>A,导致氨基酸在密码子185位点从缬氨酸变为蛋氨酸。这个变异类型在1000 Genome,ESP6500,ExAC_ALL和ExAC_EAS人群基因组数据库中出现的频率较低,Sanger证实该基因突变来源于其父亲(图9)。患者儿子基因检测也发现第2个变异c.553G>A,但眼部及全身检查未发现WS临床表型。

TU 8 Sanger sequencing测序证实外显子exon 7 c.1111G>A(p.G371S)变异类型,导致氨基酸在密码子371位点从甘氨酸变为丝氨酸;Sanger证实该基因突变来源于其母亲
a.患者母亲,b.患者父亲,c.患者,d.患者儿子。
引自:主编:.同仁眼科疑难病例精析:同仁眼科临床病例讨论会1.第1版.ISBN:978-7-117-33030-5

图 9 Sanger sequencing测序证实c.553G>A变异类型,导致氨基酸在密码子185位点从缬氨酸变为蛋氨酸;Sanger证实该基因突变来源于其父亲
a.患者母亲,b.患者父亲,c.患者,d.患者儿子。
引自:主编:.同仁眼科疑难病例精析:同仁眼科临床病例讨论会1.第1版.ISBN:978-7-117-33030-5
SIFT及Ployphen-2,Mutation Taster预测结果显示EDNRB基因这两个位点的变异都是致病变异,GEREP++预测结果显示这两个位点的变异均在保守区。
EDNRB基因是以常染色体隐性(autosomal recessive,AR)方式遗传的。测序分析显示,该患者的EDNRB基因中存在两个杂合突变,这两个突变分别来自其母亲和父亲,EDNRB基因两个位点构成了复合杂合变异。
临床表型与基因型分类
瓦登伯格综合征(WS)按照基因型可分为WS1~WS4型,而且各型依照临床表型的不同还可以进一步再分为若干亚型。WS1(OMIM: 193500)型和WS3(OMIM:148820)型常有内眦外移的表现。WS2(OMIM:193510)型则缺乏面部缺陷或内眦外移的表现。WS3(OMIM:148820)型常伴有内眦外移,上肢肌肉骨骼异常,也可称为“Klein-Waardenburg syndrome”。WS4型往往伴有慢性肠梗阻表现,WS4(OMIM: 277580) 也可称为“Shah-Waardenburg syndrome”或“Waardenburg-Hirschsprung disorder”。
瓦登伯格综合征各型与6个基因变异有关,分别是PAX3 (encoding the paired box 3 transcription factor),MITF (microphthalmia-associated transcription factor),EDN3 (endothelin 3),EDNRB (endothelin receptor type B),SOX10(encoding the Sry bOX10 transcription factor),SNAI2 (snail homolog 2)。WS4型分3个亚型,WS type 4A型和type 4B型分别与EDNRB和EDN3基因变异相关,WS type 4C型与SOX10基因变异相关。SOX10基因在神经嵴细胞的发育和移行中起重要作用,神经嵴细胞可以分化为黑素细胞,嗅鞘细胞和肠神经节神经元,故SOX10基因变异所致WS type 4C型可出现肠道发育异常,全身皮肤或头发色素脱失,神经系统发育异常。
合并开角型青光眼的机制
在本病例中,患者出生后发现双眼不对称虹膜异色,到17岁发现眼压升高,视野缩窄,诊断开角型青光眼,药物控制眼压不佳,视野损害不断进展,双眼行滤过性手术。随诊25年,眼压始终控制在12~15mmHg之间。基因检测结合临床表型,在EDNRB基因2个位点发现致病变异,诊断WS 4A型,合并开角型青光眼。这两个突变分别从其母亲和父亲遗传而来。
WS临床表型不包括青光眼,早期报道WS病例时,也曾报道过WS合并开角型青光眼、虹膜异色。1996年Kadoi和Hayasaka报道1例WS合并青光眼与视网膜静脉阻塞。Nork等报告1例WS合并双眼闭角型青光眼。Gupta等报告1例WS1型患者,WS1型与PAX3基因变异相关,在病例报告中,开角型青光眼是WS患者的重要临床特征。Abdelrahman等报告1例20岁WS患者合并青少年型青光眼。上述病例报告拓展了WS临床表型。
由于眼部的黑素细胞和小梁网组织源于神经嵴细胞,神经嵴细胞发育或移行发生异常,会导致眼部色素脱失,小梁网发育障碍。神经嵴细胞、小梁网发育异常均与青光眼发病有关,以往报告WS合并青光眼的病例,以及本病例WS合并开角型青光眼病例,患者发生青光眼的年龄均在30岁以前,与WS的基因变异导致神经嵴细胞及小梁网发育异常相关。角膜缘的间叶细胞层及小梁网,由两类成熟细胞系组成,一类起源于神经嵴,一类起源于中胚叶。WS基因变异导致颅脑神经嵴细胞发育障碍,在小梁网发育过程中,小梁薄板合并压紧的结果,影响了虹膜在发育过程中的缓慢向后移动过程,影响了房角的正常发育,导致在青少年时期房水引流障碍,发生青光眼。
EDNRB基因新位点变异类型
本病例患者及亲属的基因检测发现WS4型EDNRB基因exon 7 c.1111G>A,导致氨基酸在密码子371位点从甘氨酸变为丝氨酸,c.553G>A,导致氨基酸在密码子185位点从缬氨酸变为蛋氨酸。Sanger证实基因突变分别来源于其母亲和父亲。这两个位点的变异都是致病性变异,本病例报告首次发现与WS发病相关的EDNRB基因新的位点变异类型。
WS在临床中虽属罕见疾病,但因其特殊的临床表型和特征,也是比较容易发现的,患者基因检测可协助诊断。随着WS病例的不断发现,其临床表型也在不断拓展中。患儿特殊的皮肤及头发色素脱失、虹膜异色及内眦外移的面部特征,应引起注意是否为WS患者,肠道及神经系统,听力、肌肉骨骼系统发育异常注意检查,儿科、神经科会诊可协同诊断WS多种临床表型。
[1] PINGAULT V, ENTE D, DASTOT-LE MOAL F, et al. Review and update of mutations causing Waardenburg syndrome[J]. Hum Mutat, 2010, 31(4): 391-406.
[2] SHI Y, LI X, JU D, et al. A novel mutation of the MITF gene in a family with Waardenburg syndrome type 2: a case report[J]. Exp Ther Med, 2016, 11(4): 1516-1518.
[3] JAN I A, STROEDTER L, HAQ A U, et al. Association of Shah-Waardenburg syndrome: a review of 6 cases[J]. J Pediatr Surg, 2008, 43(4): 744-747.
[4] CHEN K, ZONG L, LIU M, et al. De novo dominant mutation of SOX10 gene in a Chinese family with Waardenburg syndrome type Ⅱ[J]. Int J Pediatr Otorhinolaryngol, 2014, 78(6): 926-929.
[5] ZAMAN A, CAPPER R, BADDOO W. Waardenburg syndrome: more common than you think![J]. Clin Otolaryngol, 2015, 40(1): 44-48.
[6] TAMAYO M L, GELVEZ N, RODRIGUEZ M, et al. Screening program for Waardenburg syndrome in Colombia: clinical definition and phenotypic variability[J]. Am J Med Genet A, 2008, 146A(8): 1026-1031.
[7] LIU X, WANG S, XING Z, et al. Targeted next-generation sequencing identified a novel variant of SOX10 in a Chinese family with Waardenburg syndrome type 2[J]. J International Medical Research, 2020, 48(11): 1-8.
[8] KUHLBRODT K, HERRBARTH B, SOCK E, et al. SOX10, a novel transcriptional modulator in glial cells[J]. J Neurosci, 1998, 18(1): 237-250.
[9] CHEN K, ZONG L, ZHAN Y, et al. Genetic counseling for a three-generation Chinese family with Waardenburg syndrome type Ⅱ associated with a rare SOX10 mutation[J]. Int J Pediatr Otorhinolaryngol, 2015, 79(5): 745-748.
[10] SHIELDS C L, NICKERSON S J, AL-DAHMASH S, et al. Waardenburg syndrome: iris and choroidal hypopigmentation: findings on anterior and posterior segment imaging[J]. JAMA Ophthalmol, 2013, 131(9): 1167-1173.
[11] ARIMOTO Y, NAMBA K, NAKANO A, et al. Chronic constipation recognized as a sign of a SOX10 mutation in a patient with Waardenburg syndrome[J]. Gene, 2014, 540(2): 258-262.
[12] KADOI C, HAYASAKA S, YAMAMOTO S. Branch retinal vein occlusion in a patient with Waardenburg syndrome[J]. Ophthalmologica, 1996, 210(6): 354-357.
[13] NORK T M, SHIHAB Z M, YOUNG R S L, et al. Pigment distribution in Waardenburg's syndrome: a new hypothesis[J]. Graefe's Arch Clin Exp Ophthalmol, 1986, 224(6): 487-492.
[14] GUPTA V, AGGARWAL H C. Open angle glaucoma as a manifestation of Waardenburg's syndrome[J]. Indian J Ophthalmol, 2000, 48(1): 49-50.
[15] ABDELRAHMAN A M, AMIN R H. Juvenile open-angle glaucoma with Waardenburg syndrome: a case report[J]. J Glaucoma, 2021, 30(1): e1-e4.









