


 收藏
收藏 已收藏
已收藏麻婧、段晓明、唐炘
色素血管性斑痣性错构瘤(phakomatosis pigmentovascularis,PPV)是一种罕见的累及全身多系统的疾病,文献报道可与GNAQ和GNA11变异有关,皮肤表现主要为皮肤鲜红斑痣合并色素痣,眼部表现主要为青光眼、脉络膜血管瘤和色素异常。PPV可与Sturge-Weber综合征(SWS)或Klipple-Trenaunay综合征(KTS)并发。现报道于本院明确诊断并接受治疗的PPV合并双侧SWS典型病例1例。
1例13岁男性患儿,因“双眼视力差10年,发现眼压高1年”,应用多种降眼压药物无效后就诊于我院,颜面部及全身皮肤可见多发斑片状色素沉积及紫红色病灶,双眼眼压增高,结膜血管迂曲扩张,巩膜色素沉着,双眼眼底色素异常增多,左眼番茄酱样改变,头颅MRI多发异常血管影,诊为“双眼青光眼合并先天异常(左眼绝对期),色素血管性斑痣性错构瘤,双侧性Sturge-Weber综合征”,给予右眼小梁切除+丝裂霉素+玻璃体腔注药术(曲安奈德2mg),术后眼压控制稳定。
青光眼;色素血管性斑痣性错构瘤;Sturge-Weber综合征
患儿男性,13岁,因“发现双眼视力差10年,眼压高1年”,就诊于我院眼科门诊。患儿自出生后即出现颜面部青紫,未予特殊诊治,10年前发现双眼视力差,就诊于当地医院,诊断为“双眼屈光不正”,给予配镜治疗,之后双眼视力逐渐下降,伴有流泪,偶伴头痛,不伴眼痛、眼红、畏光等不适,1年前体检时发现双眼眼压增高,右眼45mmHg,左眼测不出,当地医院给予“噻吗洛尔、拉坦前列腺素、布林佐胺滴眼液”治疗后,眼压控制不佳,遂就诊于我院。患儿出生时足月顺产,孕期、产时无异常,生后未吸氧,否认家族遗传病史。查体可见躯干部及四肢多发、大片浅灰蓝、暗蓝色斑片,边缘逐渐移行为正常皮肤色,符合蒙古斑表现(图1)。颜面部多发不规则暗红色斑片,为鲜红斑痣(葡萄酒色痣),左侧面部和唇部增厚隆起,鼻尖部、双耳郭、眼睑青灰色色素异常,为蒙古斑,颞侧浅白色斑片,摩擦不发红,为贫血痣(图2)。右眼视力0.01,矫正不提高,红绿色觉正常,6m光感,光定位正常,左眼视力无光感;眼压右眼21mmHg,左眼38mmHg,右眼结膜无充血,巩膜可见斑片状灰蓝色色素沉积,角膜透明,可见Haab纹,前房深,虹膜纹理清,色素明显,瞳孔圆,直径4mm,直接对光反射阳性,间接对光反射阴性,晶状体清亮,眼底色素异常增多,色暗,视盘边清色淡,上、下方盘沿明显变窄,相应处神经纤维层缺损,C/D约0.9,黄斑中心凹反光不清。左眼内斜约15°,结膜、表层巩膜血管增多、迂曲,巩膜瓷白色,角膜透明,前房深,虹膜纹理清,瞳孔圆,直接5mm,直接对光反射阴性,间接对光反射阳性,晶状体清亮,豹纹状眼底,色素异常增多,弥漫性脉络膜血管瘤番茄酱样改变,视盘苍白,C/D约1.0,黄斑中心凹反光消失,后巩膜葡萄肿(图3,图4)。眼A、B超提示双眼玻璃体混浊,左眼著,眼轴右眼31mm,左眼34.41mm。头颅MRI示右侧顶叶、双侧基底节、放射冠、侧脑室旁、脉络丛多发异常血管影,静脉畸形可能性大(图5)。患者诊断为“双眼青光眼合并先天异常,色素血管性斑痣性错构瘤,双侧性Sturge-Weber综合征,左眼内斜视”,于全麻下行右眼小梁切除+丝裂霉素+玻璃体腔注药术(曲安奈德2mg),术后眼压控制良好。

图1 蒙古斑
四肢及躯干部多发、大片浅灰蓝、暗蓝色斑片,边缘逐渐移行为正常皮肤色。
引自:主编:.同仁眼科疑难病例精析:同仁眼科临床病例讨论会1.第1版.ISBN:978-7-117-33030-5
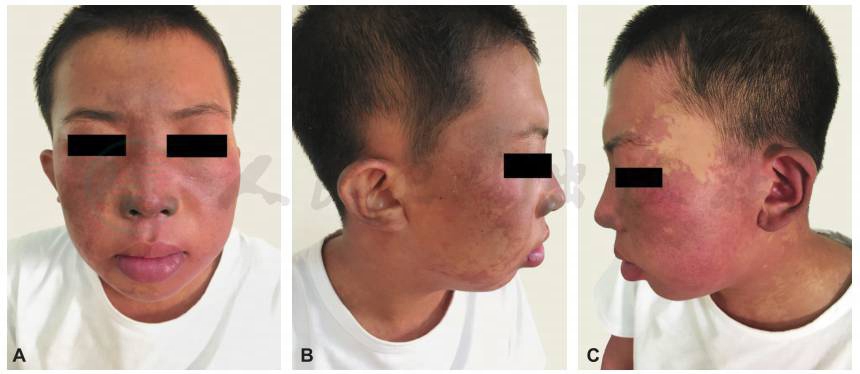
图2 头面部鲜红斑痣、蒙古斑及贫血痣
颜面部多发不规则暗红色斑片,为鲜红斑痣(葡萄酒色痣),左侧面部和唇部增厚隆起,鼻尖部、双耳郭、眼睑青灰色色素异常,为蒙古斑,颞侧浅白色斑片,摩擦不发红,为贫血痣。
引自:主编:.同仁眼科疑难病例精析:同仁眼科临床病例讨论会1.第1版.ISBN:978-7-117-33030-5
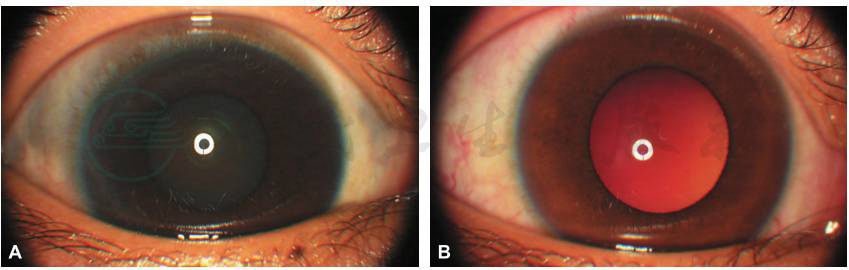
图3 双眼前节表现
A.右眼结膜无充血,巩膜可见斑片状灰蓝色色素沉积;B.左眼结膜、表层巩膜血管增多、迂曲,巩膜瓷白色,角膜透明。
引自:主编:.同仁眼科疑难病例精析:同仁眼科临床病例讨论会1.第1版.ISBN:978-7-117-33030-5
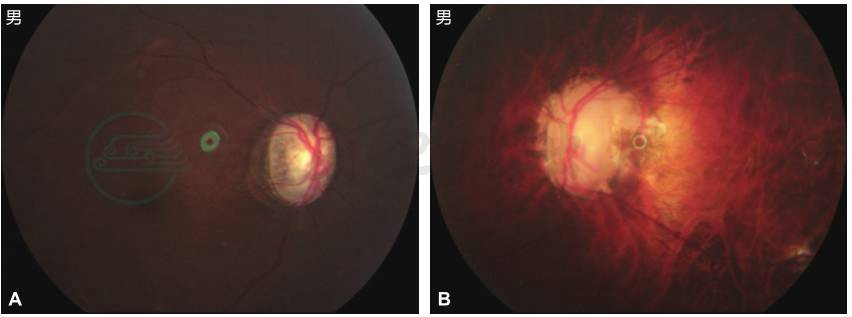
图4 双眼眼底改变
A.右眼眼底色素异常增多,色暗,视盘界清色淡,上、下方盘沿明显变窄,相应处神经纤维层缺损,C/D约0.9,黄斑中心凹反光不清;B.左眼豹纹状眼底,色素异常增多,弥漫性脉络膜血管瘤番茄酱样改变,视盘苍白,C/D约1.0,黄斑中心凹反光消失,后巩膜葡萄肿。
引自:主编:.同仁眼科疑难病例精析:同仁眼科临床病例讨论会1.第1版.ISBN:978-7-117-33030-5
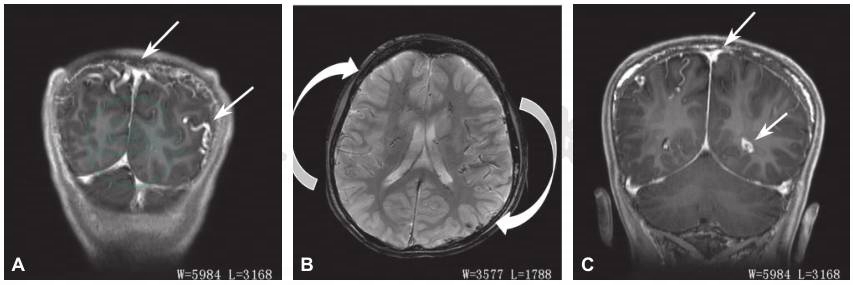
图5 头颅MRI示右侧顶叶、双侧基底节、放射冠、侧脑室旁、脉络丛多发异常血管影,静脉畸形可能性大(箭头所指范围)
引自:主编:.同仁眼科疑难病例精析:同仁眼科临床病例讨论会1.第1版.ISBN:978-7-117-33030-5
临床表现
PPV最初在1947年由Ota首次报道,表现为血管痣伴发广泛色素痣,亚洲人、非洲人多见,出生即有,与神经嵴发育异常相关。PPV分为四型,Ⅰ型(cesioflammea)表现为鲜红斑痣合并蒙古斑,Ⅱ型(spilorosea)表现为淡粉红色痣合并斑痣,Ⅲ型(cesiomarmorata)表现为先天性毛细血管扩张性大理石皮肤合并蒙古斑,Ⅳ型为其他类型,其中Ⅰ型最容易与SWS或KTS合并。SWS是一种脑三叉神经血管瘤病,发病率约1/50000~1/20000,男女无差异,90%为单侧,发病机制可能为GNAQ突变,诊断主要通过典型的面部鲜红斑痣及同侧软脑膜血管瘤。约50%合并SWS的PPV患者会合并青光眼,尤其是有眼部表现的患者。PPV合并青光眼的可能发病机制为神经嵴细胞迁移异常导致的前房角或者Schlemm管的发育异常、虹膜或睫状体的前移、高巩膜静脉压等。本例患者为PPV合并双侧性SWS的少见病例,需要与双眼先天性婴幼儿型青光眼进行鉴别。先天性婴幼儿型青光眼发病年龄早,与小梁网的发育异常有关,一般不伴有其他眼外全身异常。Klippel-Trenaunay综合征也可以表现为鲜红斑痣伴有青光眼,但同时还伴有静脉畸形、曲张以及软组织和骨的肥大异常。本例患者眼轴长,豹纹状眼底,但是存在Haab纹,提示早期眼压升高,眼轴变长为继发改变,而非原发的病理性近视。
治疗方案
药物治疗方面首选碳酸酐酶抑制剂、β受体阻滞剂,低龄儿童慎用α受体激动剂,前列腺素类药物可能通过增加巩膜葡萄膜途径流量起到一定作用。手术方面可以考虑选择性激光小梁成形术、房角切开、滤过性手术和睫状体破坏性手术。合并SWS的PPV患者进行滤过性手术时要尤其注意爆发性脉络膜上腔出血和脉络膜脱离的可能性。由于PPV的患者眼底本身可有大量色素,可能会遮蔽典型的番茄酱样眼底改变,对此应提高警惕。本例患者术中可见右眼巩膜基质层间大量色素,因此术前眼底像并无典型SWS眼底表现。在本例患者的手术治疗中,我们应用了可调节缝线来控制眼压、降低出血和脉络膜脱离的风险,并且在玻璃体腔内留置了曲安奈德以减轻炎症反应、降低脉络膜脱离的风险。部分PPV患者同时存在潜在的恶变可能,在治疗眼部及全身皮肤病损的同时,需要长期的监测和随访。
[1]ABDOLRAHIMZADEH S,PUGI DM,DE PAULA A,et al.Ocular manifestations in phakomatosis pigmentovascularis: current concepts on pathogenesis,diagnosis,and management.Surv Ophthalmol,2021,66(3):482-492.
[2]PATIL B,SINHA G,NAYAK B,et al.Bilateral sturgeweber and phakomatosis pigmentovascularis with glaucoma,an overlap syndrome.Case Rep Ophthalmol Med,2015,2015:106932.
[3]FERNANDEZ-GUARINO M,BOIXEDA P,DE LAS HERAS E,et al.Phakomatosis pigmentovascularis:clinical findings in 15 patients and review of the literature.J Am Acad Dermatol,2008,58(1):88-93.
[4]HAPPLE R.Phacomatosis pigmentovascularis revisited and reclassified.Arch Dermatol,2005,141(3):385-388.









