


 收藏
收藏 已收藏
已收藏患者男性,55岁。因“多饮、多尿9年,行走不稳1年”于2015年2月6日入院。
现病史
患者于9年前(2006年)出现多饮、多尿、烦渴,白天和夜间小便每小时1次,尿色清亮,伴皮肤干燥、体重下降、便秘、乏力。2007年因昏迷就诊于外院,实验室检查:血糖36.14mmol/L,血清钠183mmol/L,血清钾5.40mmol/L,血清氯140mmol/L;头部MRI显示垂体柄结节状增粗;禁水加压素试验结果不详,诊断为“中枢性尿崩症”,予醋酸去氨加压素片(弥凝)0.05mg(1次/8h)口服,症状控制可,尿量约1 000ml/d,未服用降糖药,血糖控制可。1年半前(2013年8月)家属发现其走路稍显不稳,曾摔倒1次。1年前(2014年2月)出现言语模糊,但可正常交流,走路不稳无明显进展,日常生活活动未受影响,可开车、游泳。8个月前(2014年6月)头部MRI显示脑干、小脑、右侧额叶、左侧颞叶多发性异常信号,增强扫描呈结节状强化;PET/CT扫描显示右侧小脑胶质瘤伴脑内多发性转移瘤可能[最大标准化摄取值(SUVmax)为14.96];胸腹盆腔CT未见明显异常;遂行脑组织活检术(活检部位为右侧脑桥臂,2014年7月1日)。活检术后患者行走不稳加重,右侧肢体活动不稳,构音障碍加重,偶有饮水呛咳,四肢肌力尚可。病理检查显示,(右侧脑桥臂)脑组织水肿,局灶性髓鞘脱失;血管壁、血管周围和脑实质内局灶性淋巴细胞和中性粒细胞浸润,淋巴细胞主要表达T细胞标记;神经胶质细胞增生,少数增生的神经胶质细胞有轻度异型性,组织学改变以炎症反应可能性大。腰椎穿刺脑脊液检查(2014年8月)压力180mmH2O,白细胞计数2×106/L,蛋白定量0.62g/L、葡萄糖3.89mmol/L、氯化物 128mmo/L,细胞学提示淋巴细胞反应;髓鞘碱性蛋白(MBP)1.21nmol/L(<0.55nmol/L),寡克隆区带(OB)阴性,抗 Hu、Yo和Ri抗体阴性。血清囊虫IgG抗体、肺吸虫IgG抗体、曼氏裂头蚴IgG抗体均阴性。临床考虑脱髓鞘病变可能,不排除肿瘤。予甲泼尼龙1000mg/d,使用5天,减量至500mg/d,使用3天,再改为泼尼松60mg/d,逐渐减量,共治疗5个月。治疗期间,患者自觉行走不稳略有改善,多次复查影像学无明显变化。复查头部MRI(2015年1月30日)显示病灶较前略有增大,为求进一步诊断与治疗,至我院就诊,门诊以“颅内多发性病变待查”收入院。患者自发病以来,无骨痛、心慌、胸闷、突眼等症状与体征,近2年双侧上眼睑和右侧背部新发黄色结节,无压痛;饮食、睡眠尚可,大小便正常,近1年体重无明显变化。
既往史、个人史及家族史
均无特殊。
入院后体格检查
体温36.5℃,脉搏78次/min,呼吸12次/min,血压132/86mmHg。双侧上眼睑外侧、右侧背部“豆粒”样黄色扁平结节,无压痛。神志清楚,构音障碍。粗测双眼视力可,双侧瞳孔等大、等圆,直径约3mm,对光反射灵敏,眼球活动正常,双眼凝视可见水平眼震,无复视。双眼闭目有力,右侧鼻唇沟浅,右侧口角低,伸舌居中。软腭活动度可,咽反射存在。右侧肢体肌力5-级、左侧5级,肌张力均正常,右侧肱二头肌反射、膝腱反射略高于左侧,右侧Babinski征可疑阳性。右侧指鼻试验和跟-膝-胫试验欠稳准,可见意向性震颤,右侧反击征阳性。深浅感觉正常。脑膜刺激征阴性。行走不稳,步基宽,直线行走不能。
辅助检查
实验室检查血、尿、粪便常规、肝肾功能试验、凝血功能均正常。血清甘油三酯(TG)3.92mmol/L,总胆固醇(TC)、低密度脂蛋白胆固醇(LDL-C)、高密度脂蛋白胆固醇(HDL-C)正常。血清超敏C反应蛋白(hsCRP)5.36mg/L(0~3mg/L),红细胞沉降率(ESR)24mm/h,抗核抗体(ANA)谱、抗中性粒细胞胞质抗体(ANCA)、抗可提取性核抗原(ENA)抗体、水通道蛋白4(AQP4)抗体、血管紧张素转换酶(ACE)均阴性。CD8+T细胞异常激活。血清抗巨细胞病毒IgG和IgM阳性、Ⅰ型和Ⅱ型单纯疱疹病毒IgG阳性。巨细胞病毒DNA和pp65抗原均阴性。结核分枝杆菌抗体、结核感染T细胞斑点试验(T-SPOT.TB)、布鲁氏菌凝集试验、莱姆病毒抗体、1,3-β-D葡聚糖检测(G试验)、隐球菌抗原、人类免疫缺陷病毒(HIV)抗体、梅毒螺旋体明胶凝集试验(TPPA)均阴性。空腹血糖6.60mmol/L,糖化血红蛋白(HbA1c)正常。促肾上腺皮质激素(ACTH)12.12pmol/L(参考值范围0~10pmol/L),皮质醇、生长激素(GH)、胰岛素样生长因子1(IGF-1)均正常。血清睾酮5.86nmol/L(参考值范围6.07~27.27nmol/L),卵泡刺激素(FSH)、黄体生成素(LH)、泌乳素(PRL)、雌二醇、孕激素均正常。肿瘤标志物筛查未见明显异常,血清抗Hu、Yo和Ri抗体阴性。腰椎穿刺脑脊液检查压力135mmH2O,白细胞计数2×106/L,蛋白定量0.67g/L、葡萄糖4.20mmol/L、氯化物123mmol/L,细胞学未见异常,乳酸2.47mmol/L;EB病毒和巨细胞病毒DNA、快速血浆反应素试验(RPR)、细菌和真菌涂片等均阴性;甲胎蛋白(AFP)、癌胚抗原(CEA)阴性;寡克隆区带、AQP4抗体阴性。MRI显示垂体后叶高信号消失,增强扫描下丘脑和垂体柄增粗强化;左侧颞叶、右侧顶叶病灶基本同前,小脑、脑干病灶较前增大(图1)。胸部高分辨率CT可见右肺下叶结节;右肺上叶、左肺下叶少许斑片状和索条状高密度影;左侧胸膜局部增厚。99Tcm-亚甲基二磷酸盐(99Tcm-MDP)骨显像显示双侧股骨下段、胫骨两端放射性摄取对称性增高,不排除非朗格汉斯细胞组织细胞增生症中的Erdheim-Chester病;左侧上颌骨、右侧第5~9前肋、左侧第6和第9前肋放射性摄取增高,考虑骨折可能;余未见明显异常(图2)。下肢长骨X线显示双侧胫骨下段骨密度异常。超声心动图未见明显异常。副鼻窦CT显示右侧上颌窦黏膜下囊肿;双侧上颌窦和筛窦炎;左侧中鼻道狭窄。复查18F-FDG PET/CT显示右侧小脑蚓部旁、脑桥背侧偏左、右侧额叶半卵圆中心代谢增高(SUVmax为12.10),考虑恶性病变,建议行进一步检查;右侧小脑弥漫性代谢降低,考虑继发改变;双侧上颌窦炎,右侧上颌窦囊肿(图3)。

图1 头部MRI检查所见
a.横断面FLAIR成像(2014年6月)显示脑干、小脑多发性高信号;b.横断面FLAIR成像(2015年1月)显示病灶较前增大;c.横断面增强T1WI显示病灶呈结节状强化;d.矢状位T1WI显示垂体后叶高信号消失;e.冠状位增强T1WI显示垂体柄结节状增粗、强化
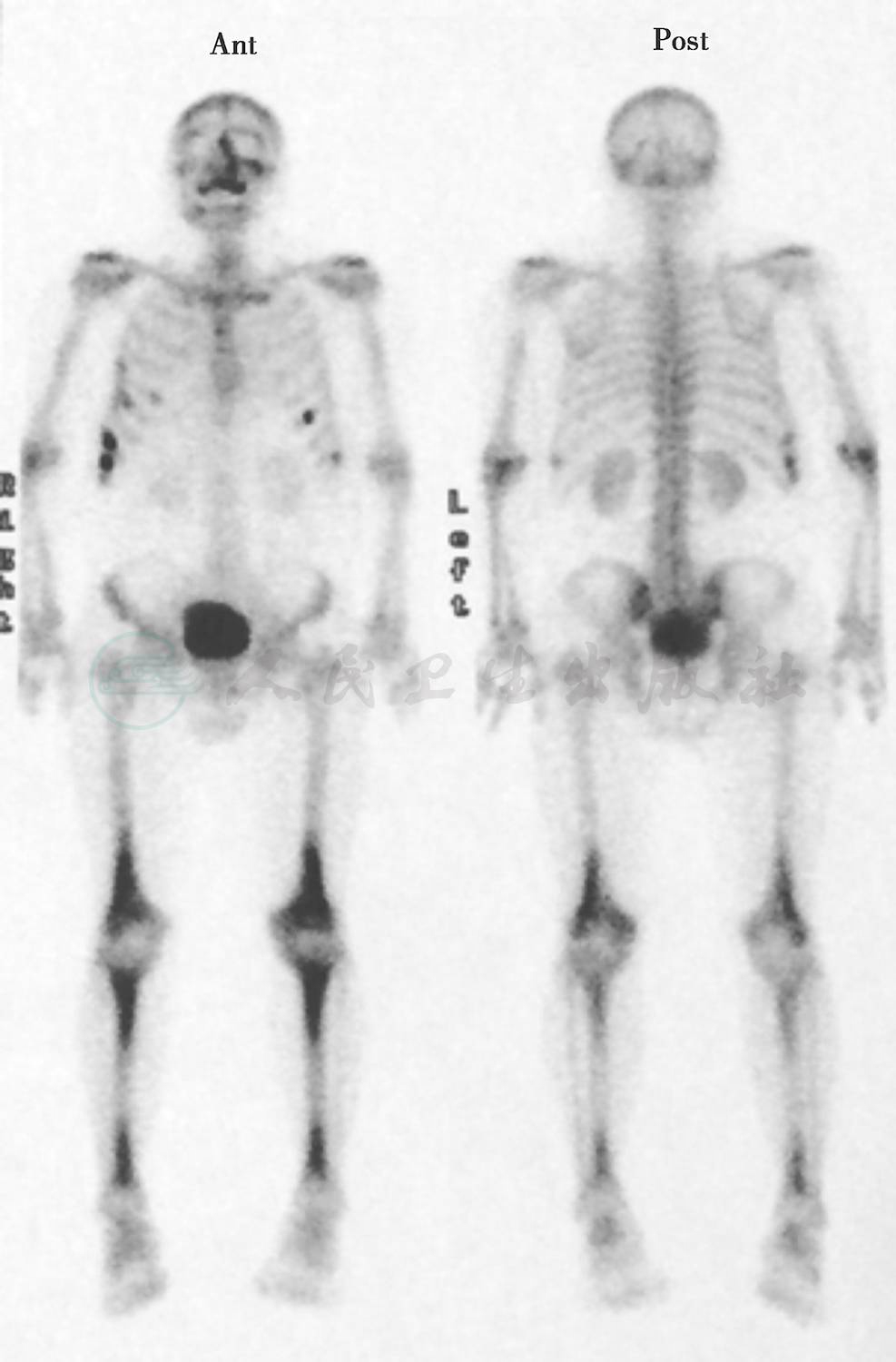
图2 99Tcm-MDP骨显像
显示双侧股骨下段、胫骨两端放射性摄取对称性增高

图3 患者头部18F-FDG PET/CT扫描
可见脑干病灶代谢增高(SUVmax=12.10)
诊断与治疗经过
(右侧脑桥臂)脑组织活检经病理科会诊,可见增生的肥胖性星形胶质细胞,小血管炎伴血管周围炎,组织细胞浸润,可见灶性髓鞘脱失。患者近2年出现眼睑和右侧背部结节,经皮肤科会诊,考虑黄瘤病,扁平黄瘤或结节黄瘤不确定。建议行皮肤组织活检术,考虑神经系统病变可能与黄瘤病相关。右侧背部结节活检提示,表皮皮突趋于消失,真皮中下部弥漫性组织细胞浸润,周围可见多核巨细胞,组织细胞纤维化明显;免疫组织化学染色,CD68阳性,CD1a、S-100蛋白、CD207阴性。左侧眼睑结节活检提示慢性炎症反应,可见大量泡沫细胞和Touton巨细胞聚集,伴慢性炎性细胞浸润;免疫组织化学染色,广谱细胞角蛋白(pCK)、CD15、CD1a、结蛋白(Des)阴性,CD34和CD68阳性,溶菌酶(lysozyme)和S-100蛋白可疑阳性。基因检测显示BRAF V600E突变。根据患者临床症状、影像学表现、皮肤活检和基因检测结果,最终诊断为非朗格汉斯细胞组织细胞增生症中的Erdheim-Chester病。经血液科会诊后予干扰素-α(IFN-α)6×106U(1次/d)皮下注射,同时继续服用醋酸去氨加压素片控制尿崩症。出院后2个月随访时,出现双下肢疼痛,可忍耐,拄拐不能独立行走。
神经科主治医师
患者中年男性,隐匿起病,慢性病程,临床主要表现为三方面:①9年前多饮、多尿、烦渴,诊断为中枢性尿崩症,口服醋酸去氨加压素片症状控制可;②近1年出现行走不稳、构音障碍,病情进展相对缓慢,大剂量甲泼尼龙冲击和泼尼松口服治疗5个月后,临床症状和影像学改善不明显;③近2年双侧上眼睑和右侧背部新发黄色结节。定位诊断:①行走不稳,构音障碍,右侧指鼻试验和跟-膝-胫试验欠稳准,可见意向性震颤,反击征阳性,水平眼震,定位于小脑及其联系纤维,以右侧显著。②双侧闭目有力,右侧鼻唇沟浅,右侧口角低,考虑右侧中枢性面瘫,定位于面神经核以上的左侧皮质脑干束;右侧肱二头肌反射、膝腱反射略高于左侧,右侧Babinski征可疑阳性,定位于左侧皮质脊髓束。结合影像学,上述症状与体征可以用脑干和小脑病变解释。③临床表现为多饮、多尿、烦渴症状,影像学显示下丘脑和垂体柄增粗、强化,提示下丘脑-垂体系统受累。④新发皮肤结节,存在皮肤受累。⑤无骨痛主诉,但99Tcm-MDP骨显像和下肢长骨X线检查提示长骨受累,为骨质硬化性表现,非破坏性改变。定性诊断:患者中年男性,隐匿起病,慢性病程,无发热、头痛、恶心、呕吐等症状。影像学表现为颅内幕上和幕下多发性病灶,以脑干和小脑病变显著,MRI呈长T1、长T2信号,周围脑组织水肿,增强扫描呈结节状强化,PET/CT显示病灶呈高代谢。影像学表现较重而临床症状较轻,进展相对较慢,同时有下丘脑-垂体系统、皮肤和骨骼受累,定性诊断考虑如下疾病:①肿瘤,脑组织活检未发现明确肿瘤证据,仅见个别神经胶质细胞异型性,但18F-FDG PET/CT显示多发性高代谢病灶,不能排除肿瘤,脑组织活检结果阴性不除外与取材部位有关,但病程较长、进展相对缓慢为不支持点。②系统性疾病,除脑实质内多发性病变外,尚有下丘脑-垂体系统、皮肤和骨骼受累,因此需考虑系统性疾病。肉芽肿性多血管炎(又称韦格纳肉芽肿)和结节病可累及皮肤和中枢神经系统,以硬脑膜受累常见,也可累及鞍区和脑实质,但该例患者未见肺部病变、激素治疗无效、血清ANCA和血管紧张素转换酶呈阴性为不支持点。鞍区和颅内多发性病变的鉴别诊断还应考虑组织细胞增生性病变,除中枢神经系统病变,组织细胞增生性病变可以累及多个系统,但仅根据临床表现和影像学鉴别诊断困难,明确诊断需组织活检和特殊染色。该例患者皮肤活检显示,CD68阳性,S-100可疑阳性,CD1a和CD207阴性,结合多系统受累表现,诊断考虑非朗格汉斯细胞组织细胞增生症中的Erdheim-Chester病。鉴别诊断:①炎性脱髓鞘病变,脑组织活检提示髓鞘脱失,炎症反应可能性大,应考虑中枢神经系统炎性脱髓鞘疾病,但该例患者颅内病灶分散,颞叶和顶叶病灶并非典型脱髓鞘病变部位,且存在脑实质病变1年余,病灶仍明显强化伴周围脑组织水肿,糖皮质激素冲击治疗后临床症状和影像学表现无改善,为不支持点。②感染,该例患者无发热、头痛,脑脊液蛋白定量稍高,呈轻度淋巴细胞反应,脑实质多发病灶,并非常见细菌、寄生虫感染,需慎重除外特殊类型病原菌。完善脑脊液细菌、真菌、病毒、寄生虫等筛查,无明确感染证据。
神经科教授
患者中年男性,尿崩症9年,近1年出现行走不稳、言语模糊,体格检查显示小脑性共济失调,头部MRI显示颅内多发病灶,以小脑和脑干为主,呈长T1、长T2信号,增强扫描可见结节状强化,PET/CT可见放射性摄取增高,结合患者眼睑和背部结节、骨扫描下肢长骨两端放射性摄取增高、皮肤活检和BRAFV600E基因突变,诊断为Erdheim-Chester病。脑组织活检显示神经胶质细胞原纤维酸性蛋白(GFAP)阳性,提示神经胶质增生,而CD68阴性,无明确支持Erdheim-Chester病的证据,但亦不能排除,因为此种病理改变可见于多种疾病。非朗格汉斯细胞组织细胞增生症可以累及鞍区,病变范围广泛,明确诊断需依据临床表现和病理学检查。该例患者颅内多发病变,占位效应相对较轻,呈相对良性病程,故不支持肿瘤。MRI显示左侧上颌窦病变,性质未定,结合文献报道Erdheim-Chester病可累及副鼻窦,经耳鼻咽喉头颈外科会诊,考虑炎症可能,但不排除原发病累及副鼻窦。因无明确病理学证据,上颌窦病变性质待定,需随诊观察。Erdheim-Chester病与朗格汉斯细胞组织细胞增生症临床表现有相似之处,需进行鉴别,后者以颅面骨、四肢骨近端、骨盆、肩胛骨受累多见,免疫组织化学染色,CD68、CD1a、S-100、CD207阳性可资鉴别。患者目前无心血管、肺部和腹膜后病变,颅内受累为Erdheim-Chester病预后不良的危险因素,糖皮质激素治疗无效,可予干扰素-α治疗。
Erdheim-Chester病(Erdheim-Chester disease,ECD)
Erdheim-Chester病(Erdheim-Chester disease,ECD)是一种罕见的非朗格汉斯细胞组织细胞增生症(NLCH),由Jacob Erdheim和William Chester于1930年首次报告。近10年来,随着人们对该病的认识增加,文献报道的病例数急骤增多。该病的平均诊断年龄约55岁,男性多于女性,约占73%。Erdheim-Chester病可以引起多系统受累,患者表现为骨痛、突眼、尿崩症、黄色瘤、肺部病变、腹膜后纤维化、中枢神经系统和心血管系统病变等。
1.发病机制
Erdheim-Chester病细胞起源尚不明确,有文献报道,Erdheim-Chester病和朗格汉斯细胞组织细胞增生症(LCH)可以并存,约12%的Erdheim-Chester病患者合并朗格汉斯细胞组织细胞增生症。细胞因子在Erdheim-Chester病发病机制中起重要作用,炎性因子和趋化因子参与组织细胞激活和募集。Arnaud等研究发现,Erdheim-Chester病患者干扰素-α,白细胞介素(IL)-12、IL-4和 IL-7,单核细胞趋化蛋白-1(MCP-1)表达水平异常升高,说明炎症反应可能在发病过程中起重要作用。此外,Erdheim-Chester病存在BRAF V600E基因突变,表明该病是一种克隆性、肿瘤性疾病。BRAF V600E突变是原癌基因BRAF的活化型突变,可见于多种肿瘤。RAS-RAF-丝裂原活化蛋白激酶/细胞外信号调节激酶(MEK)-细胞外信号调节激酶(ERK)信号转导通路在肿瘤发生发展中起重要作用,BRAF基因突变导致该通路非依赖性RAS异常激活。BRAF基因抑制剂已用于Erdheim-Chester病的治疗。
2.临床表现及影像学特点
Erdheim-Chester病临床表现多样,可累及多器官,与组织细胞增生性病变的其他类型,如朗格汉斯细胞组织细胞增生症、Rosai-Dorfman病(Rosai-Dorfmandisease,RDD)等鉴别诊断困难。①骨骼:骨骼受累最常见,主要累及四肢长骨,表现为对称性骨质硬化。所有患者均可见长骨受累,但仅50%患者有骨痛表现。朗格汉斯细胞组织细胞增生症则主要累及颅骨、四肢近端骨、骨盆和肩胛骨。Erdheim-Chester病放射性核素骨显像表现为长骨干骺端对称性放射性核素聚集;X线表现为骨主干和干骺端骨质硬化。②中枢神经系统:25%~50%患者存在中枢神经系统病变,可累及脑实质和脑膜。脑实质病变见于脑桥、齿状核和大脑半球,MRI表现为病灶强化征象,应注意与原发性肿瘤和继发性转移瘤、脱髓鞘病变及炎症相鉴别。脑膜受累应注意与脑膜炎、肉芽肿性病变、Rosai-Dofman病相鉴别。中枢神经系统受累提示预后不良,是死亡结局的独立危险因素,故应进行基线MRI评价。③皮肤:常见的皮肤损害是眼睑黄斑瘤,面部、颈部、躯干、腹股沟和腋窝也可见黄色或棕红色斑块。仅从皮肤损害形态很难与幼年性黄色肉芽肿(juvenile xanthogranuloma,JXG)相鉴别,后者多系统受累少见。④内分泌系统:约25%患者有尿崩症表现,也可表现为高泌乳素血症、促性腺激素缺乏、血清睾酮降低等内分泌功能异常。影像学可见腺垂体、垂体柄、下丘脑受累,部分患者可见上述结构受累但并无内分泌功能异常。⑤肺部:超过50%的患者影像学表现为肺部受累,病变主要位于肺实质和胸膜,多无临床症状,少数表现为咳嗽和呼吸困难。高分辨率CT可见肺叶间隔增厚、肺组织“毛玻璃”样影,单纯肺实质病变不常见。⑥眼部:约25%患者有单眼或双眼浸润性病变,眶内占位表现为突眼,严重者可出现眼肌麻痹和视力下降。应注意与炎性假瘤、Graves病、肉芽肿性病变、淋巴瘤等鉴别。⑦心血管系统:心血管系统受累常见但多无临床症状,约2/3患者累及胸主动脉或腹主动脉,形成“coated aorta”(包被型主动脉)典型影像学改变;肾动脉受累可出现肾性高血压。心脏病变包括右心房假瘤样病变(pseudotumor)、心包纤维化、心脏瓣膜浸润,心脏受累是重要死因。⑧腹膜后浸润:约30%患者影像学可见腹膜后浸润表现,肾脏周围受累可出现“hairy kidney”(毛发肾)的典型征象,也可引起肾积水、输尿管狭窄。腹膜后浸润与腹膜后纤维化不同,盆腔输尿管和下腔静脉多不受累。
3.诊断
Erdheim-Chester病的诊断主要依靠特征性的组织病理学及临床和影像学表现。①组织病理学:泡沫细胞(foamy cell)或富含脂质的组织细胞(lipid-laden histiocyte)浸润,通常可见Touton巨细胞,病变周围可见纤维化。免疫组织化学染色,CD68、CD163、凝血因子 XⅢa阳性,CD1a、Langerin阴性,S-100阴性或弱阳性。②影像学检查:下肢长骨骨干和干骺端对称性骨质硬化常见,99Tcm-MDP骨显像可见长骨远端放射性高摄取,X线可见骨质硬化表现。PET/CT敏感性低于放射性骨显像,但可发现骨骼以外其他器官受累,是评估Erdheim-Chester病疾病负荷的重要方法。CT和MRI也可见骨骼受累,但X线检查可能漏诊。此外,CT所示肾脏周围脂肪浸润形成的“hairy kidney”也具有特异性。
值得注意的是,即使临床和影像学表现典型,也应行组织活检以明确诊断并行BRAF基因检测。Haroche等研究发现,约54.17%(13/24)Erdheim-Chester病患者BRAFV600E基因突变。Cangi等改进检测方法,采用锁核酸聚合酶链反应联合焦磷酸测序技术,检出所有患者(18/18)均BRAF V600E基因突变。
4.治疗
Erdheim-Chester病临床罕见,前瞻性研究少,目前尚无随机对照临床试验。①干扰素-α:目前,支持证据较多的是干扰素-α和聚乙二醇干扰素-α(PEG-IFN-α)。一项纳入53例Erdheim-Chester病患者的前瞻性非随机观察性队列研究显示,行干扰素-α或PEG-IFN-α治疗的46例患者预后改善。根据疾病严重程度和器官受累情况确定治疗剂量,标准剂量是:干扰素-α 3×106U(3次 /周)或 PEG-IFN-α 135µg(1次 /周)。对于中枢神经系统受累或心脏受累的患者可予大剂量干扰素治疗[干扰素-α(6~9)×106U(3次/周)或PEG-IFN-α 180µg(1次/周)]。关于最佳治疗疗程尚不确定,一项纳入24例行干扰素-α治疗的高危Erdheim-Chester病患者的临床研究显示,63.64%(7/11)中枢神经系统受累和78.57%(11/14)心脏受累患者病情稳定或改善。②其他:丝氨酸/苏氨酸蛋白激酶抑制剂vemurafenib(维莫非尼)已应用于BRAF V600E基因突变患者,其临床和影像学表现均显著改善。细胞因子抑制剂阿那白滞素(anakinra)、英利昔单抗(infliximab)、托珠单抗(tocilizumab)等也已应用于Erdheim-Chester病的治疗,但鉴于病例数较少,部分药物仍处于临床试验阶段,药物疗效尚待进一步评价。糖皮质激素可以减轻颅内病灶周围水肿,但单独治疗无效,目前研究西罗莫司联合泼尼松疗效的临床试验正在进行中。
5.预后
Erdheim-Chester病患者预后不良,受累器官越多、预后越差,中枢神经系统受累是不良预后的独立危险因素。1996年的一项纳入59例Erdheim-Chester病患者的研究显示,平均随访32个月,仅1/3患者生存。晚期研究显示,干扰素治疗的5年生存率约为68%。
综上所述,该例患者以尿崩症发病,数年后出现皮肤黄色瘤及脑干、小脑和大脑半球多发病灶,结合影像学上骨硬化表现、皮肤活检病理结果及BRAFV600E基因阳性突变,Erdheim-Chester病诊断明确。患者目前有中枢神经系统、内分泌系统、皮肤和骨骼受累,尚无心血管系统损害和腹膜后浸润表现。Erdheim-Chester病的明确诊断主要依靠组织活检,但活检部位的选择和组织病理学的诊断具有挑战性。病变组织并非总表现为典型泡沫细胞浸润,部分受累器官病变表现为合并纤维化的非特异性炎症反应,或仅表现为纤维化而组织细胞少见,给明确诊断增加了难度。该例患者脑组织活检、组织病理学并未呈现典型Erdheim-Chester病表现,因此,多部位组织活检可以提高诊断准确性。该例患者存在中神经系统受累,予较大剂量干扰素-α治疗后症状仍进行性加重,可能提示预后不良。
[1]Arnaud L,Hervier B,Neel A,et al.Cns involvement and treatment with interferon-alpha are independent prognostic factors in erdheim-chester disease:A multicenter survival analysis of 53 patients. Blood,2011117(10):2778-2782.
[2]Arnaud L,Gorochov G,Charlotte F,et al.Systemic perturbation of cytokine and chemokine networks in erdheim-chester disease:A single-center series of 37 patients. Blood,2011,117(10):2783-2790.
[3]Haroche J, Charlotte F, Arnaud L, et al. High prevalence of braf v600e mutations in erdheim-chester disease but not in other non-langerhans cell histiocytoses.Blood,2012,120(13):2700-2703.
[4]Davies H,Bignell GR,Cox C,et al.Mutations of the braf gene in human cancer.Nature,2002,417(6892):949-954.
[5]Drier A,Haroche J,Savatovsky J,et al.Cerebral,facial,and orbital involvement in erdheim-chester disease:Ct and mr imaging findings.Radiology,2010,255(2):586-594.
[6]Diamond EL,Dagna L,Hyman DM,et al.Consensus guidelines for the diagnosis and clinical management of erdheim-chester disease. Blood,2014,124(4):483-492.
[7]Braiteh F,Boxrud C,Esmaeli B,et al.Successful treatment of erdheim-chester disease,a non-langerhans-cell histiocytosis,with interferon-alpha.Blood,2005,106(9):2992-2994.
[8]翟菲菲,乔雷,钱敏,等.多饮多尿9年行走不稳伴言语不清1年.中国现代神经疾病杂志,2015,15(8):681-687.









