


 收藏
收藏 已收藏
已收藏患者女性,19岁。因“发作性四肢无力10年”入院。
现病史
患者入院10年前剧烈运动后出现双下肢无力,可平路行走,不可跑步、蹲起,上肢力量可。否认肢体麻木、走路不稳等。无晨轻暮重现象。当地医院查血钾正常低限,血清肌酸激酶(creatine kinase,CK)升高(200~300U/L),症状持续1周自行缓解。此后,类似症状发作1次。7年前患者运动后出现四肢无力,无法站立,上臂抬举困难。当时查体:双下肢肌力3+级,双上肢肌力5级,四肢感觉正常,腱反射对称引出。实验室检查:血钾正常、CK 221U/L;诊断为“周期性麻痹”,给予补钾和营养神经治疗无效。症状持续10天后自行缓解。以后上述症状反复发作,常于月经来潮10天前剧烈运动后出现,持续约10天至月经来潮前自行缓解,发作频率为2个月1次至1个月2次。发作期间反复查血钾正常。曾怀疑“正常血钾型周期性麻痹”,查相关基因正常,予乙酰唑胺、氯化钾治疗无效。为进一步诊治就诊于我院。患者自起病以来精神、食欲、睡眠可,大小便正常,体重无明显改变。既往、个人、月经和婚育史无殊。母亲于妊娠前曾有反复发作性肢体无力,补钾后症状可缓解,产后症状未再发。表姐有室性心律失常病史。
入院后体格检查
发育正常,手脚偏小,颈蹼,第5足趾侧弯。心、肺、腹部检查无明显异常。神清语利,高级智能粗测无明显异常。脑神经检查正常。四肢肌力5级,肌张力正常;四肢腱反射对称活跃;病理征未引出。共济运动和深浅感觉检查正常。
入院后辅助检查
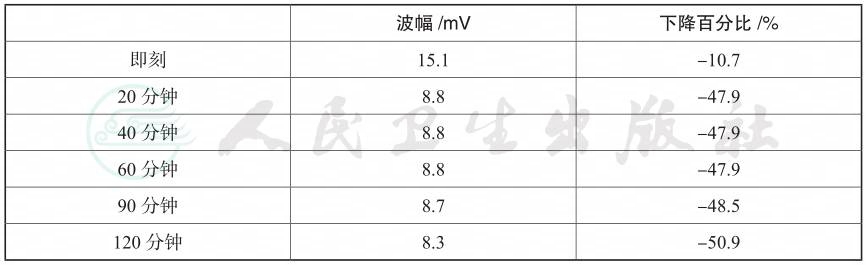
常规检查,血、尿、粪便常规、肝肾全和凝血功能正常。代谢与内分泌相关:糖化血红蛋白正常,空腹胰岛素17.90µIU/ml;甲状腺功能正常;甲状旁腺激素正常;血镁正常;24小时尿儿茶酚胺正常;血总皮质醇正常;血ACTH正常;1mg地塞米松过夜试验可被抑制;24小时尿钾正常;立位醛固酮正常;血管紧张素Ⅱ275.03pg/ml(参考值范围25.3~145.3);肾素正常;性激素:睾酮0.77ng/ml(参考值范围0.1~0.75);硫酸脱氢表雄酮338.5µg/dl(参考值范围51~321);β人绒毛膜促性腺激素正常。心脏方面检查:心肌酶正常;心电图正常;超声心动图正常。电生理检查:肌电图和神经传导速度正常;运动诱发试验异常(表1)。基因检查:血钾相关周期性麻痹基因筛查正常。
表1 运动诱发试验(右侧尺神经CMAP)
波幅/mV |
下降百分比/% |
|
即刻 |
15.1 |
-10.7 |
20分钟 |
8.8 |
-47.9 |
40分钟 |
8.8 |
-47.9 |
60分钟 |
8.8 |
-47.9 |
90分钟 |
8.7 |
-48.5 |
120分钟 |
8.3 |
-50.9 |
入院后诊断与治疗
入院后患者爬两层楼梯后出现发作。查体:左下肢肌力4级,右下肢肌力5-级,双上肢肌力5级,四肢肌张力正常,病理征阴性。心电图示窦性心律,频发室早(室性早搏),校正的Q-T间期(QTc)399~438ms,可见u波(图1)。血钾、血糖和血镁正常。结合患者特殊外貌和发作时的心电图异常,行Andersen-Tawil综合征相关基因筛查,发现基因 KCNJ2 突变(+),c.199C>T(p.Arg67Trp),考虑患者诊断 Andersen-Tawil综合征明确,转入心内科进行心律失常的评估和治疗。
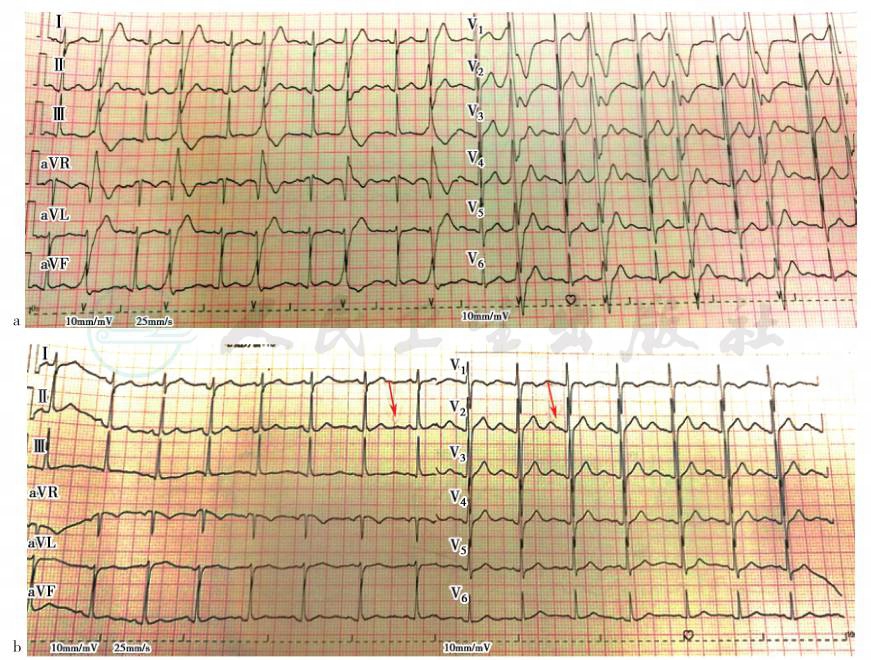
图2 患者发作期心电图
a.室早二联律;b.发作期心电图可见u波
神经科主治医师
患者青年女性,儿童期起病,反复发作四肢无力,仅有运动系统受累,无感觉系统异常。患者发作频率逐渐增加,由之前的2个月1次、1个月1次,到近期1个月2次;发作期时间延长,早期发作1周后可缓解,近期需要2周至1个月方可缓解;主要诱发因素为剧烈运动,另外,患者发作与月经周期也有密切相关性,常为月经前10天发作,10天后在月经前期好转,不排除与性激素相关;发作期多次查血钾正常。家族史:患者母亲曾在妊娠前有多次发作性肢体无力病史,考虑为“周期性麻痹”,发作时有心悸症状;患者表姐有多次室性心律失常发作史。发作间期神经系统查体未见明显异常;发作期左下肢肌力4级,右下肢肌力5-级,双上肢肌力5级;四肢腱反射低,病理征阴性;另外患者外貌有先天性疾病的表现,如手脚小、颈蹼、第5足趾侧弯等。定位诊断:根据患者发作期仅有肢体无力,无感觉障碍,病理征阴性,血清CK升高的特点考虑病变定位于肌肉。定性诊断:患者基因检测提示Andersen-Tawil综合征。该例患者从发病到确诊经历了相对漫长的过程。根据患者发作性四肢弛缓性瘫痪,血钾正常或偏低的特点,首先考虑为“周期性麻痹”,曾经行血钾相关周期性麻痹基因检查正常。入院后依据患者特殊外貌特征,考虑有无先天遗传性疾病,儿科会诊提示有Noonan综合征可能,但该病与目前与周期性麻痹无明确相关性。最终,一次发作后发现患者有明显的心律失常,心电图示室早二联律,有u波出现。结合患者周期性麻痹、室性心律失常、外貌畸形,查阅文献后考虑Andersen-Tawil综合征可能性大。基因检查后确诊。
心内科主治医师
患者青少年女性,幼年起病。临床主要表现为周期性麻痹、心脏受累以及发育异常。反复行心电图检查见频发多源室性早搏、u波,未见QTc间期明显延长。结合患者母亲、表姐多年室性早搏、QTc间期延长等表现,基因检查KCNJ2突变(+),诊断方面考虑Andersen-Tawil综合征诊断明确。患者已行运动平板试验检查,但患者室性心律起搏位置不一,射频消融较难消除其室性早搏心律。分析其Holter心电图,心室率快多出现于晨起后,考虑与交感兴奋相关,建议加用美托洛尔12.5mg,1次/12h治疗。患者无晕厥或室速发作,暂无植入ICD(植入型心律转复除颤器)指征。Anderson-Tawil综合征预后相对较好,恶性心律失常,如室速、室颤等发生率较其他先天性长Q-T间期综合征为低,嘱患者及家属如有晕厥发作,及时就诊。继续比索洛尔控制室性心律失常治疗。
Andersen-Tawil综合征(Andersen-Tawil syndrome)
Andersen-Tawil综合征是一种遗传性的临床综合征。其临床表现三联征包括周期性麻痹、室性心律失常或长Q-T间期,以及特定的外貌异常。患者一般在10~20岁发病。将近60%的患者有完整的三联征,而超过80%的患者拥有三联征中的至少两项。患者的发作性无力症状可自发出现,也经常在剧烈运动后的休息过程中发作。随着发作次数逐渐增多,许多患者在发作间期也常遗留有一定程度的近端肌无力。患者室性心律失常,主要包括多源性室早、多形性室速、双向性室速,其他的心电图异常包括显著的u波、Q-T间期延长等。心脏方面患者可表现为无症状,也可仅有心悸,较少出现晕厥、心搏骤停以及猝死等严重症状。一项法国的36名Andersen-Tawil综合征患者的回顾性研究表明,在长达9.5年的时间里,有4例患者出现过晕厥,1例患者出现过心搏骤停,但没有患者死亡。有报道显示一些Andersen-Tawil综合征患者可出现扩张型心肌病。患者的外貌异常包括宽额头、眼裂狭小、眼距增宽、宽鼻梁、球状鼻、低位耳、小下颌、薄上唇、三角形脸等特征的面容异常,乳牙不退、少齿、牙列拥挤等牙齿畸形,以及小手小脚、第五指弯曲、第二、三指并指等骨骼畸形。此外,患者认知功能可有轻度受累,主要包括学习能力、执行功能与抽象能力异常。
检查方面,Andersen-Tawil综合征患者血钾水平在发作期可以升高、正常、降低,但以降低为主。发作间期常规的电生理检查,如NCV通常是正常的。但更进一步的电生理检查,如运动诱发试验,则可发现运动后CMAP的波幅下降超过40%。心电图及Holter心电图的异常表现,与上述相同。
本例患者临床上以正常血钾周期性麻痹为主,偶有低血钾周期性麻痹。在周期性麻痹发作期,我们监测到患者频繁发作室性心律失常,以室早二联律最为显著(图1)。另外,也偶有可疑u波出现。然而我们在患者的心电图中并未发现Q-T间期延长。患者有明确的颈蹼、小手小脚、第五趾弯曲等表现,提示有先天遗传病可能。在电生理检查方面,发作间期时患者的NCV、EMG检查均正常,但运动诱发试验表现为运动诱发后右侧尺神经CMAP波幅降低45%~50%(表1)。
目前Andersen-Tawil综合征的临床诊断需要有≥2个典型临床表现,或是仅有1个典型临床表现,但亲属有≥2个典型临床表现。本例患者三个典型临床表现均存在,临床上可诊断为Andersen-Tawil综合征。
Andersen-Tawil综合征的最终确诊依赖于基因检查,该病为常染色体显性遗传,接近60%临床诊断为Andersen-Tawil综合征的患者有基因KCNJ2的变异。KCNJ2是编码内向整流钾通道2蛋白(Kir2.1)的基因,因此这个基因突变导致钾离子通道正常结构与功能的破坏,或是使钾离子通道不能被正常插入细胞膜上,导致骨骼肌及心肌上钾离子流动异常,最终导致出现周期性麻痹与心律失常。这种KCNJ2基因变异的Andersen-Tawil综合征被称为1型Andersen-Tawil综合征。另有40%临床诊断为Andersen-Tawil综合征的患者未发现KCNJ2基因突变,目前病因尚不明确,被称为2型Andersen-Tawil综合征。有病例报道一例临床诊断为Andersen-Tawil综合征的患者,基因检测发现KCNJ5变异,因此,发现更多与Andersen-Tawil综合征相关基因是未来研究方向之一。
50%Andersen-Tawil综合征患者的突变基因来自父母,50%患者则是由于基因直接突变所致。在一个KCNJ2基因的p.Arg67Trp突变家系中,发现只有男性患者发生周期性麻痹,女性患者仅有心脏病变,但男女患者均可发生外貌异常。本例患者在进行基因检查后发现有KCNJ2基因的p.Arg67Trp杂合突变,故可确诊为1型Andersen-Tawil综合征。但与上述文献不同之处在于,本例患者为女性,却同时拥有周期性麻痹、室性心律失常和外貌异常三个典型表现。目前患者父母也完成了相关基因检查,其父亲的基因型正常,母亲为杂合突变。依据患者母亲年轻时曾有周期性麻痹表现,以及患者母亲家族中一位患者的表姐同样有室性心律失常的家族史,患者母亲家族很可能为Andersen-Tawil综合征的遗传家族。
Andersen-Tawil综合征治疗较复杂,对于周期性麻痹发作患者,若血钾低于正常,应给予补钾治疗直至血钾水平正常;若血钾水平为正常低限,则应将其调整至正常高限;若血钾水平较高,可适当补糖增加钾离子向细胞内转运。在周期性麻痹发作间期,减少发作频率与严重程度是治疗的主要目标,主要药物为碳酸酐酶抑制剂和缓释钾。
心脏治疗方面,快速性心律失常致晕厥者,可植入心脏除颤器。严重频繁室性心律失常者,可经验性应用氟卡尼,以延缓左心室功能下降。此外,Andersen-Tawil综合征患者应慎用抗心律失常药物,特别是I类抗心律失常药物,因为它们有可能增加神经系统无力症状的发作。对于一些已知可以延长Q-T间期的药物亦应避免使用,而噻嗪类和非保钾类利尿剂,由于可造成人为的低血钾,也应避免使用。即使患者没有发作,也应每年行心电图及Holter心电图检查以监测心律失常情况。
对于本例患者,以正常血钾周期性麻痹为主要表现,应适当补钾以维持血钾水平至正常上限。此外,患者室性心律失常有晕厥、心搏骤停及猝死风险,应在心内科随诊,给予抗心律失常治疗。患者若有妊娠要求,建议行产前基因诊断,以降低后代出现遗传病的风险。
综上所述,本例患者以周期性麻痹为主要临床表现,在之前的10年病程中及本次住院前期,均以低钾型周期性麻痹或正常血钾型周期性麻痹为主要诊断方向,但基因检查均无任何发现。患者特殊的外貌特征,让我们怀疑患者是否有儿科相关先天遗传病。最终在患者周期性麻痹发作时,通过心电图发现患者有显著的室性心律失常,最后基因检查确诊Andersen-Tawil综合征。因此,对于周期性麻痹患者,我们需要关注患者有无特殊外貌、发作期是否有心律失常,以及其他家庭成员有无类似发作和心律失常病史,以减少Andersen-Tawil综合征的漏诊。
[1]Kukla P,Biernacka EK,Baranchuk A,et al.Electrocardiogram in Andersen-Tawil syndrome.New electrocardiographic criteria for diagnosis of type-1 Andersen-Tawil syndrome.Current Cardiology Reviews,2014,10 (3):222-228.
[2]Yoon G,Quitania L,Kramer JH,et al.Andersen-Tawil syndrome:definition of a neurocognitive phenotype. Neurology,2006,66(11):1703-1710.
[3]Davies NP,Imbrici P,Fialho D,et al.Andersen-Tawil syndrome:new potassium channel mutations and possible phenotypic variation. Neurology,2005,65(7):1083-1089.
[4]Kokunai Y,Nakata T,Furuta M,et al.A Kir3.4 mutation causes Andersen-Tawil syndrome by an inhibitory effect on Kir2.1.Neurology,2014,82(12):1058-1064.
[5]Airey KJ,Etheridge SP,Tawil R,et al.Resuscitated sudden cardiac death in Andersen-Tawil syndrome.Heart Rhythm,2009,6(12):1814-1817.
[6]张梦雨,徐雁,沈建中,等.发作四肢无力10年,中国现代神经疾病杂志,2017,17(7):546-549.









