


 收藏
收藏 已收藏
已收藏患者男性,49岁。因“记忆力减退1年、加重伴视物异常3个月”于2013年12月11日入院。
现病史
患者自2012年12月开始“经常找不到常用物品”,此后逐渐出现找不到常去地点,伴焦虑、注意力不集中,视物模糊、反应迟钝;约6个月后上述症状明显加重且伴视野缩小、视物发白、视物变形和听力明显减退。遂至当地医院就诊,腰椎穿刺脑脊液各项指标均于正常值范围(具体不详);血清抗中性粒细胞胞质抗体-抗丝氨酸蛋白酶(PR3-ANCA)395U/L(<20U/L),考虑为ANCA相关性血管炎,并自2013年10月22日起接受糖皮质激素(具体不详)治疗(静脉滴注),1周后改为泼尼松50mg(1次/d)口服,连续治疗3周症状无缓解并出现不认识自己家和周围环境等症状,遂于2013年11月27日至我院神经内科就诊,以“认知功能减退”收入院进一步明确诊断与治疗。病程中无幻觉、妄想、人格改变,无肢体麻木、无力、肢体抽搐,无恶心、呕吐、腹痛、腹泻、皮肤色素沉着。
既往史
无特殊。
个人史
无特殊。
家族史
育有一子,家族成员中无类似表现,无家族遗传病病史。
入院后体格检查
头发稀疏;心、肺、腹未见明显异常。高级智能检查,神清语利,但不能准确报出年、月、日,不能读表,不知自己3年前、3年后年龄,不能计算11减3,但会写自己姓名。脑神经检测粗测双眼视力下降,双耳听力减退,其余未见明显异常。运动系统检查,四肢肌张力正常、四肢肌力5级;双侧膝腱反射对称亢进,其余腱反射对称活跃,双侧踝阵挛阳性,双侧Babinski征阳性;感觉系统中肢体轻触觉、针刺觉、音叉振动觉基本正常,图形觉、实体觉减退;共济运动、步态大致正常。
入院后诊断和治疗经过
入院后完善认知功能评价,各项神经心理学量表评分为简易智能状态检查量表(MMSE)15分、蒙特利尔认知评价量表(MoCA)9分。实验室检查:血、尿、粪便常规,肝肾功能试验和血清电解质测值均于正常值范围;感染4项呈阴性反应,血清红细胞沉降率、超敏C反应蛋白正常;风湿免疫性疾病相关指标免疫球蛋白、补体正常,血清抗中性粒细胞胞质抗体谱PR3-ANCA 66RU/ml,其余各项均于正常值范围,抗可提取性核抗原(ENA)抗体谱和抗核抗体谱阴性。头部MRI提示双侧顶枕颞叶、胼胝体压部及锥体束走行区对称性异常信号,符合肾上腺脑白质营养不良表现(图1)。遂完善肾上腺脑白质营养不良相关检查:极长链脂肪酸C22 26.98mg/L、C24 44.53mg/L、C26 1.45mg/L、C24/C22 1.65、C26/C22 0.05。ABCD1基因检测显示ABCD1基因第1号外显子呈小片段缺失,TACCTTCGTCAACAGTGC 432-449 del导致145~150位氨基酸缺失(图2)。内分泌相关检查:血浆ACTH(8:00am)125.2pmol/L(参考值范围0~10.1pmol/L)、总皮质醇(8:00am)421.7nmol/L(参考值范围110.6~616.4nmol/L),24小时尿皮质醇123.9nmol/24h(参考值范围33.9~285.7nmol/24h),性腺轴激素硫酸脱氢表雄酮(DS)1.8μmol/L(参考值范围1.2~8.6μmol/L)、雌二醇(E2)12.5pmol/L(参考值范围73.0~175.8pmol/L)、睾酮(T)9.9nmol/L(参考值范围 13.4~23.6nmol/L)、FSH 5.6IU/L(参考值范围1.4~18.1IU/L)、LH 6.85IU/L(参考值范围1.5~9.3 IU/L)、孕酮(P)0.73nmol/L。临床考虑肾上腺脑白质营养不良。入院后未予特殊治疗。
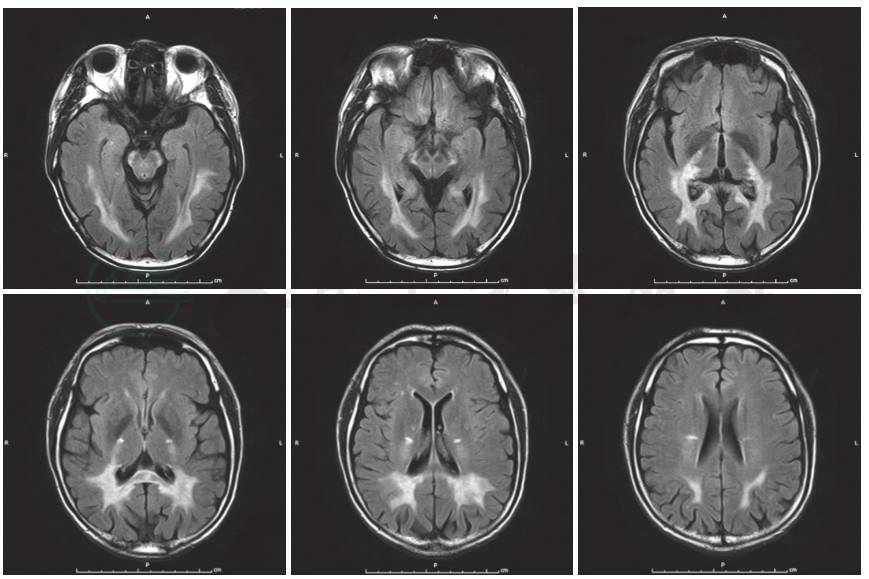
图1 患者头部MRI检查
FLAIR显示双侧顶枕颞叶、胼胝体压部及锥体束走行区对称性异常,呈FLAIR高信号
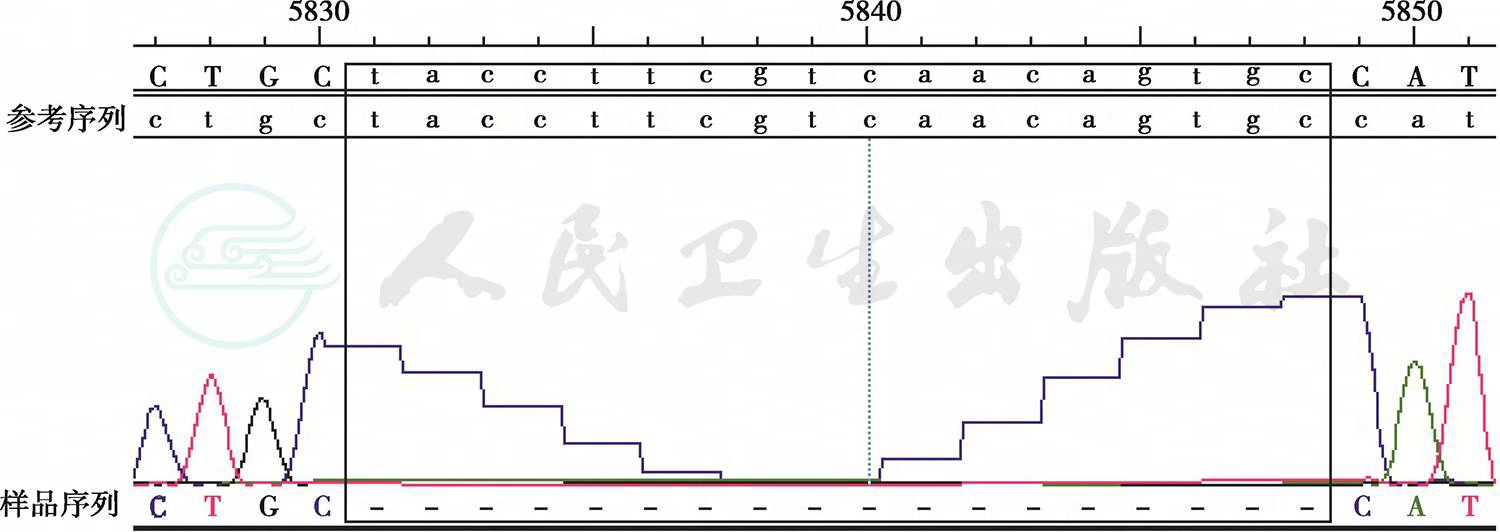
图2 患者基因测序检查结果
可见黑框所示缺失片段
神经科主治医师
患者为中年男性,呈慢性病程。临床表现为记忆力减退、反应迟钝等认知功能减退,伴视物异常。既往无殊,家族成员中无类似表现。体格检查远、近记忆力减退,计算力、理解力减退;粗测双眼视力差、双耳听力差,其余脑神经无明显异常;四肢肌力5级、肌张力正常;双侧膝腱反射亢进、踝阵挛阳性、双下肢锥体束征阳性;轻触觉、针刺觉、音叉振动觉大致正常,四肢图形觉、实体觉减退;共济运动、步态基本正常。头部MRI提示双侧顶枕颞叶、胼胝体压部及锥体束走行区对称异常信号。血清极长链脂肪酸检测C26、C24/C22和C26/C22水平显著升高;基因检测为ABCD1基因突变。定位诊断:①该患者有记忆力减退、反应迟钝、理解力下降等认知功能减退表现,结合头部MRI所示脑白质异常信号,主要定位于皮质下白质;②双下肢膝腱反射亢进、双侧踝阵挛阳性、双下肢病理征阳性,结合头部MRI双侧锥体束走行区异常信号,定位于双侧锥体束;③患者视物异常、视力下降,结合头部MRI双侧枕叶白质广泛异常信号,定位于双侧视觉传导通路。定性诊断:该患者主要表现为认知功能减退、双侧锥体束受累;MRI显示双侧后部白质、胼胝体压部和锥体束走行区呈对称性长T2异常信号;血清极长链脂肪酸C26、C24/C22与C26/C22显著升高;ABCD1基因突变;考虑X连锁型肾上腺脑白质营养不良(成人脑型)诊断。多次血清PR3-ANCA检测均呈阳性,需警惕合并系统性血管炎如韦格纳肉芽肿。
内分泌科医师
患者无恶心、呕吐、腹泻、皮肤色素沉着等肾上腺皮质功能减退表现;实验室检查血清ACTH 570.0pg/ml、总皮质醇15.39μg/dl、24小时尿皮质醇44.94μg/24h。根据上述检查,可诊断艾迪生(Addison)病,但暂不考虑行糖皮质激素替代治疗,若出现手术、创伤等应激情况需采取激素替代治疗,随访观察食欲、体重、血压、电解质等项指标的变化。
风湿免疫科医师
一般认为,血清PR3-ANCA阳性诊断韦格纳肉芽肿的特异性可达95%。但该例患者无上、下呼吸道受累症状,无血管事件,无血尿,且血清肌酐水平于正常值范围,红细胞沉降率、血清超敏C反应蛋白正常,抗可溶性核抗原(ENA)抗体谱、抗核抗体谱阴性,考虑结缔组织病、血管炎之证据不足。可请眼科医生进行眼底检查,以排除眼底血管炎改变。
眼科医师
患者双眼前节未见明显异常,眼底无血管炎相关表现。
神经科教授
结合患者认知功能、视力听力减退等症状,双侧锥体束征,MRI显示双侧后部白质、胼胝体压部及锥体束走行区对称性长T2异常信号,血清极长链脂肪酸水平显著升高、ABCD1基因突变合并艾迪生病,诊断主要考虑X连锁型肾上腺脑白质营养不良(成人脑型)。对于已存在的认知功能减退,目前尚无特殊治疗。可向患者及家属进行遗传咨询,其育有一子,X连锁型肾上腺脑白质营养不良是一种X连锁隐性遗传性疾病,患病男性,不会将致病基因传给其子。
X连锁型肾上腺脑白质营养不良(X-linked adrenoleukodystrophy,X-ALD)
X连锁型肾上腺脑白质营养不良为临床常见过氧化物酶体病,呈X连锁隐性遗传。致病基因为ABCD1,其突变导致过氧化物酶体中β氧化障碍,引起极长链脂肪酸在各种组织蓄积。该病常累及肾上腺皮质与神经系统,神经系统受累后可表现为炎性脱髓鞘(脑型)或累及脊髓传导束的慢性进行性轴索病变(肾上腺脊髓神经病型)。
X连锁型肾上腺脑白质营养不良临床表现复杂多样,主要包括脑型、肾上腺脊髓神经病型、艾迪生病型和无症状型。以男性好发、女性携带者偶可受累。脑型X连锁型肾上腺脑白质营养不良主要表现为认知功能减退,发病年龄为4~8岁,成年人亦可发病。X连锁型肾上腺脑白质营养不良以患者体内生化改变和基因异常为典型表现:①生化学改变,饱和极长链脂肪酸蓄积,特别是二十六烷(C26:0)与二十四烷(C24:0)酸增加。既往研究显示,半合子突变患者极长链脂肪酸水平(均值±标准差)为C26:0(1.18±0.53)μg/ml,C26:0/C22:0(0.07±0.04)和C24:0/C22:0(1.49±0.45);而正常成年男性为C26:0(0.29±0.29)μg/ml,C26:0/C22 :0(0.01±0.01)、C24:0/C22 :0(0.85±0.17)。本例患者C26为1.45mg/L,C26/C22 0.05、C24/C22 1.65,支持X连锁型肾上腺脑白质营养不良之诊断。②基因异常,致病基因ABCD1的改变。本例患者ABCD1基因第1号外显子小片段缺失,TACCTTCGTCAACAGTGC 432-449 del,导致145~150位氨基酸缺失。该突变既往未曾报道,但既往研究发现错义突变致第148位氨基酸改变为致病突变,故而认为其第145~150位氨基酸缺失为致病突变,X连锁型肾上腺脑白质营养不良(成人脑型)诊断明确。
X连锁型肾上腺脑白质营养不良具有特征性的头部MRI表现为脑白质对称性长T1、长T2信号,可累及胼胝体及脑干;病变由后向前发展逐一累及枕、顶、颞、额叶;增强后病灶周围区域强化,呈“蝴蝶”状。该例患者MRI可见头后部白质对称性长T1、长T2信号,支持诊断。
X连锁型肾上腺脑白质营养不良的治疗以对症支持治疗为主,尚无特异性治疗,目前治疗方法包括:①激素替代治疗。对于伴有肾上腺皮质功能不全的X连锁型肾上腺脑白质营养不良患者均需行肾上腺皮质激素替代治疗,本例病例无肾上腺皮质功能不全表现,但储备功能不足,在应激情况下需补充糖皮质激素。②Lorenzo油饮食治疗。Lorenzo油是三油酸甘油酯(glyceryl trioleate)和三芥酸甘油酯(glyceryl trierucate)按4:1制成的混合物。口服Lorenzo油并配合适当低脂饮食,可使患者血浆极长链脂肪酸在4周之内降至正常。然而,众多临床研究显示,Lorenzo油不能改变已经出现症状的X连锁型肾上腺脑白质营养不良患者的症状与体征,特别是脑型患者的疾病进程。③造血干细胞移植治疗。造血干细胞移植是目前治疗早期儿童脑型X-ALD最有效的方法,对于晚期成年人作用有限。因此目前本例患者尚无特殊治疗方法。
[1]Moser HW,Mahmood A,Raymond GV.X-linked adrenoleukodystrophy. Nat Clin Pract Neurol,2007,3 (3):140-151.
[2]Moser HW.Adrenoleukodystrophy:phenotype,genetics,pathogenesis and therapy.Brain,1997,120(Pt 8):1485-1508.
[3]Engelen M,Kemp S,de Visser M,et al.X-linked adrenoleukodystrophy(X-ALD):clinical presentation and guidelines for diagnosis,follow-up and management.Orphanet J Rare Dis,2012,7:51.
[4]Moser AB,Kreiter N,Bezman L,et al.Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls.Ann Neurol,1999,45(1):100-110.
[5]Fuchs S,Sarde CO,Wedemann H,et al.Missense mutations are frequent in the gene for X-chromosomal adrenoleukodystrophy(ALD).Hum Mol Genet,1994,3(10):1903-1905.
[6]Shimozawa N,Honda A,Kajiwara N,et al.X-linked adrenoleukodystrophy:diagnostic and follow-up system in Japan. J Hum Genet,2011,56(2):106-109.
[7]范思远,倪俊,杨荫昌,等.记忆力减退1年加重伴视物异常3个月.中国现代神经疾病杂志,2014,14(8):738-740.









