


 收藏
收藏 已收藏
已收藏患者男性,46岁。因“关节疼痛9个月,记忆力减退3个月”于2007年6月15日入院诊治。
现病史
9个月前(2006年10月)患者无任何原因出现全身大关节疼痛,非对称性,部位不固定,外院实验室检查“红细胞沉降率增快,血清C反应蛋白水平升高”,但抗核抗体等项免疫学指标均呈阴性,考虑为“反应性关节炎”,经对症治疗后病情好转。8个月前患者出现双眼红肿、胀痛并伴轻微头痛,但无流泪、畏光等症状,当地医院考虑为“结膜炎”,予地塞米松5mg/d,静脉滴注,3天后改为口服泼尼松10mg/d口服,关节疼痛症状减轻,但双眼红肿无改善并呈渐进性加重,由于“眼压较高”考虑“巩膜炎”而将口服泼尼松剂量增至40mg/d,治疗7天后双眼红肿症状有所好转,泼尼松口服剂量逐渐减至10mg/d维持治疗。其间患者腹部出现环形红色斑疹,无瘙痒、脱屑,并逐渐扩展至胸部、背部、颜面部和四肢,伴瘙痒,3~4天后未予治疗逐渐消退,遗留色素沉着。4个月前(2007年2月)无明显诱因患者出现活动时双手抖动,但无持物不稳;3个月后(2007年3月)出现午后持续发热,体温最高达38.5℃,伴乏力及头部持续性胀痛、记忆力减退(以近记忆力减退为主),并两次在夜间出现“谵妄”。外院头部MRI检查显示双侧岛叶、海马回、双侧基底节区及半卵圆区多个小片状长T1、长T2信号,增强扫描无强化(图1)。腰椎穿刺检查脑脊液压力为210mmH2O,白细胞计数42×106/L,单个核细胞70%,多核细胞30%,脑脊液蛋白0.630g/L,葡萄糖和氯化物正常;抗酸和墨汁染色,脑脊液细胞学未见肿瘤细胞,脑脊液细菌涂片等均无异常发现。血白细胞计数16×109/L、中性粒细胞比例0.91,结核分枝杆菌抗体(TB-Ab)阴性,EB病毒抗体(EBV-Ab)检测呈阳性反应。考虑“病毒性脑炎”给予更昔洛韦、头孢类抗生素(具体剂量不详)、地塞米松10mg/d静脉滴注,治疗10天后症状明显好转出院,但记忆力无改善,仍以口服泼尼松10mg/d维持治疗。出院后约2周患者再次出现低热,体温约为37.5℃,伴乏力,咳嗽、咳痰,双手关节游走性疼痛,伴红、热但无肿胀,同时出现“谵妄”。当地医院实验室检查显示:血常规,白细胞计数14.90×109/L,中性粒细胞比例0.84;红细胞沉降率20mm/h,结核菌素纯蛋白衍生物(PPD)皮肤试验呈阴性反应,抗可提取性核抗原(ENA)抗体、免疫球蛋白定量、蛋白电泳和甲状腺功能检测均于正常范围;腰椎穿刺检查脑脊液压力为130mmH2O,白细胞计数2×106/L,蛋白定量0.64g/L。脑电图检查未见明显异常;头部MRI扫描病灶依旧存在,无强化表现,提示脑内异常信号,炎性或血管炎性改变。遂按照“病毒性脑炎”对症治疗,病情好转后出院。出院后继续口服激素泼尼松7.50mg/d,约1周后患者再次出现“谵妄”、行走不稳,以及右耳红肿、疼痛并进行性加重情况,不伴发热,遂来我院就诊。病程中无声嘶、喘息、喉及气管软骨前压痛,无鼻塌陷及疼痛等症状与体征,无光过敏、雷诺现象、口腔溃疡及外阴溃疡等现象。患者自发病以来兴趣减退,入睡困难。大小便正常。体重无明显变化。
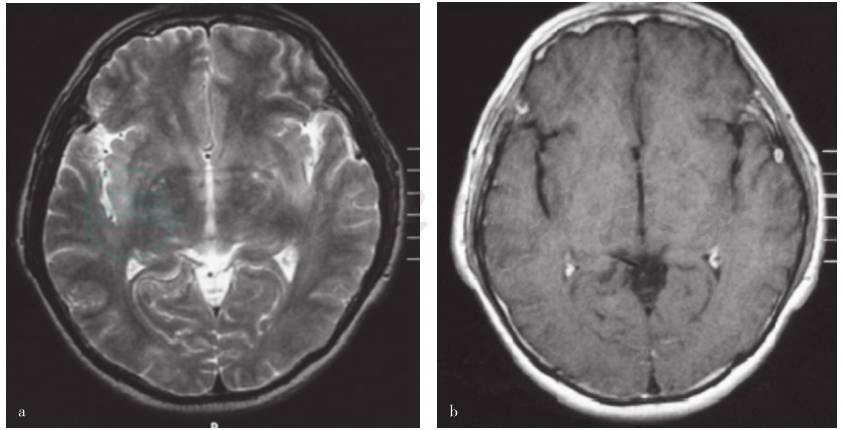
图1 患者头部MRI检查
a.横断面T2WI序列可见双侧基底节区,额颞叶皮质及皮质下多发散在长T2信号,边界模糊,病灶新旧不等;b.增强扫描无强化
既往史、个人史、家族史
无特殊。
入院后体格检查
体温37.4℃,脉搏82次/min,呼吸18次/min,BP 140/100mmHg。颈部、前胸、手足均可见陈旧性色素沉着。左眼内侧结膜可见0.50cm溃疡,无脓性分泌物。右耳耳郭发红但触之无明显疼痛。神经系统检查神志清晰,语速较快。时间、地点定向力、理解力、计算力欠佳,人物定向力尚可,远近记忆力均较差。舌肌可见震颤,其余脑神经检查无异常发现。四肢肌力5级,双侧上肢肌张力呈“齿轮”样增高,双侧下肢肌张力稍高;双手、双侧下肢不自主抖动。双侧趾关节位置觉减退,复合觉减退。共济运动试验尚可,四肢腱反射对称引出,病理征阴性。脑膜刺激征阴性。
辅助检查
血常规检查白细胞计数为11.82×109/L,中性粒细胞比例0.75,血红蛋白129g/L,血小板计数400×109/L;红细胞沉降率41mm/h,C反应蛋白47mg/L;血清乳酸、梅毒快速血浆反应素试验(RPR)、EB病毒抗体和结核菌素纯蛋白衍生物皮肤试验均呈阴性。甲状腺功能、肿瘤标志物 CA 系列[包括CA199,CA50,CA242,甲胎蛋白(AFP),癌胚抗原(CEA)等]和肺癌标志物筛查基本于正常值范围。血清抗脑组织抗体(抗Hu、Yo、Ri抗体)呈阴性反应;抗中性粒细胞胞质抗体(ANCA)、抗核抗体、抗双链DNA抗体(dsDNA)、抗可提取性核抗原抗体、类风湿因子(RF)、抗心磷脂抗体(ACA),以及补体C3、C4、免疫球蛋白及血清免疫电泳检测均正常;淋巴细胞表型分析CD4/CD8细胞比例未见异常;人类白细胞抗原B27(HLA-B27)呈阴性反应。腰椎穿刺脑脊液检查压力为170mmH2O,脑脊液常规:白细胞计数8×106/L,蛋白0.75g/L,葡萄糖3.90mmol/L,氯化物117mmol/L;脑脊液细胞学结果:淋巴细胞比例95%,符合以淋巴细胞主的炎性反应;24小时免疫球蛋白合成率正常,未见寡克隆区带(OCB),髓鞘碱性蛋白(MBP)10.98nmol/L。心电图、心脏超声、经颅多普勒超声(TCD)、双侧颈动脉和椎动脉彩色超声均未见明显异常。肺CT平扫及增强扫描未发现明显异常影像。头部MRI检查,弥散加权成像(DWI)和增强扫描均显示双侧额叶皮质及皮质下、双侧基底节区、双侧侧脑室旁、内侧颞叶斑片状长T2异常信号(图2),较外院影像无明显改变。核素扫描全身骨显像无异常。脑电图报告呈边缘状态。简易智能状态检查量表(MMSE)评分为17分(异常)。耳鼻咽喉科会诊:双侧轻度传导性听力下降,考虑双侧分泌性中耳炎。与患者及家属沟通后行耳郭组织活检。
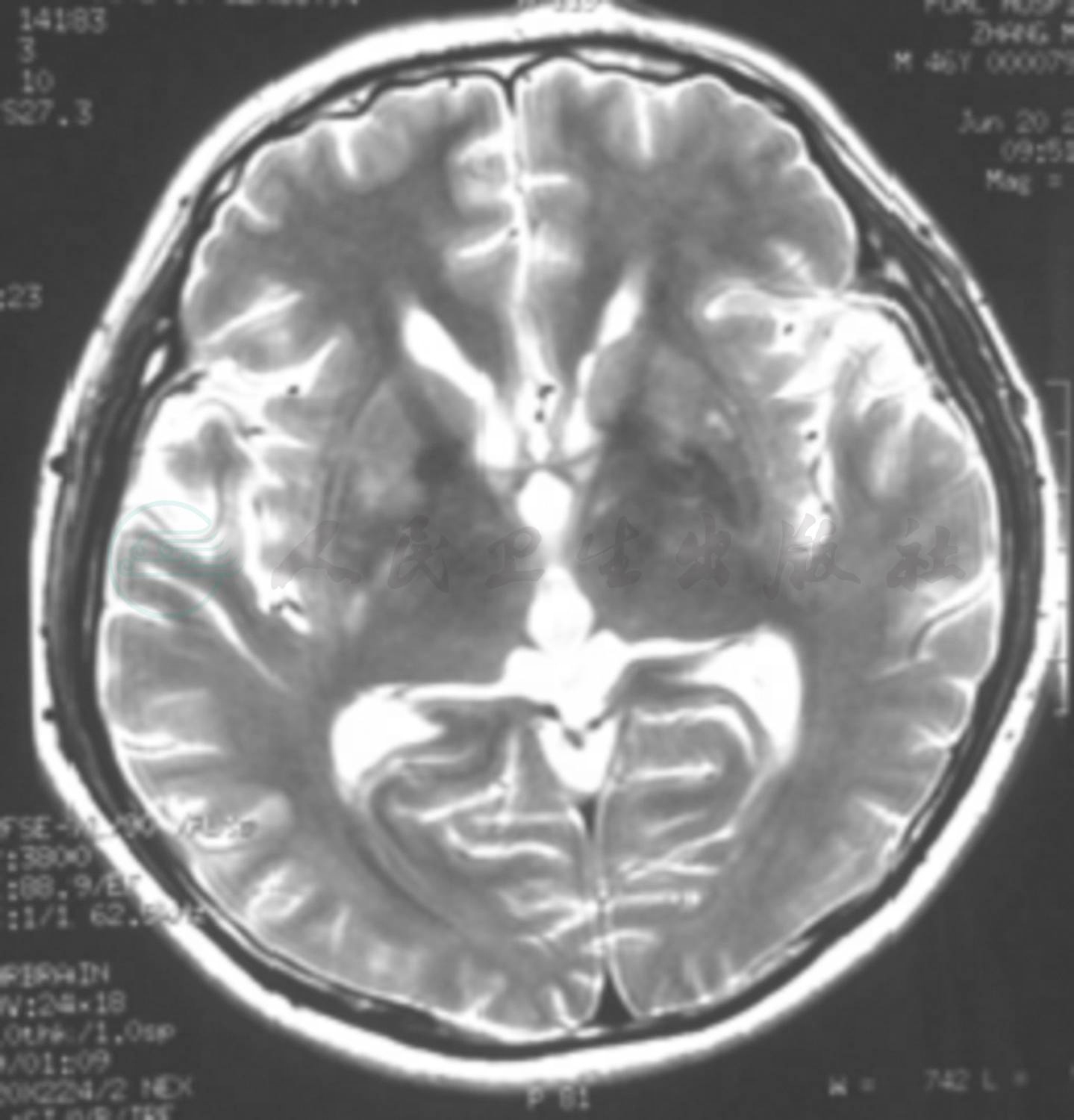
图2 患者头部MRI检查
横断面T2WI显示双侧额叶皮质及皮质下、侧脑室
神经科医师
该患者为中年男性,急性发病,亚急性病程,病情呈波动性变化,发病初期主要表现为系统性疾病症状,曾在其他医院先后被诊断为“反应性关节炎”“巩膜炎”和“右耳郭软骨膜炎”等,就诊期间还出现皮肤损害。近4个月来反复发热及神经系统症状,如头痛、发热、记忆力减退和精神行为异常等症状。病程中及入院后实验室检查白细胞计数、红细胞沉降率及血清C反应蛋白水平等炎性指标均显著升高;多次脑脊液检查提示白细胞计数、蛋白定量轻度升高,符合淋巴细胞炎性反应。头部MRI扫描显示双侧额叶皮质下、双侧基底节区、双侧侧脑室旁、内侧颞叶斑片状长T2异常信号。定位诊断:可将患者记忆力减退、精神行为异常和高级智能活动下降(定向力、记忆力、计算力等)等症状定位于双侧额颞叶;而四肢肌张力增高、不自主运动则定位在双侧基底节锥体外系。结合影像学检查病灶所在脑区,可以解释患者目前的临床症状及体征。另外,对于全身多系统损害表现,包括皮肤陈旧性色素沉着、眼结膜溃疡、耳郭发红等,考虑全身系统性受累。定性诊断:首先应考虑中枢神经系统炎症性疾病,包括感染性及非感染性炎症。根据患者亚急性病程,临床表现为发热、头痛、记忆力减退、精神行为异常等,首先,应排除病毒性脑炎的可能。但患者病程较长,约6个月,且病情反复波动,经抗病毒治疗病情仍有反复并呈渐进性加重,说明抗病毒治疗效果欠佳,此亦不符合病毒感染自限性之特点。其次,该患者影像学呈现颅内广泛性病灶,对于灰、白质并无太多的选择性,与单纯疱疹病毒性脑炎的典型灰质受累、额颞叶为主的出血坏死影像学图像不相符;同时入院后脑电图检查亦未发现典型的颞叶或以颞叶为中心累及额叶的周期性放电,因此均不支持病毒性脑炎的典型表现。对于非感染性炎性疾病方面,应注意与中枢神经系统炎性脱髓鞘性疾病急性播散性脑脊髓炎(ADEM)相鉴别。由于该患者的病程及影像学表现与急性播散性脑脊髓炎的急性发病、单相病程、白质受累为主和发病前多有感染或疫苗接种史不符,故以上两种疾病均不支持。目前,患者有多系统及脏器受累的临床表现,且先于神经系统症状出现,支持其原发病为全身系统性疾病而神经系统损害仅为其相应表现的诊断。鉴于患者在整个病程中先后出现眼、耳、关节等多器官或系统受损,并以软骨炎为特异性表现同时伴发中枢神经系统损害,故应首先考虑复发性多软骨炎伴中枢神经系统炎性病变;建议进行耳郭病理活检证实。
免疫科医师
患者为中年男性,亚急性发病,病程6个月余,病情反复;临床主要表现为关节疼痛,巩膜炎,皮疹,右耳郭软骨膜炎,听力减退、耳鸣及神经系统损害等多系统受累。实验室检查红细胞沉降率和血清C反应蛋白升高,免疫学、肿瘤标志物及感染指标均呈阴性,脑脊液检测提示慢性炎性改变;头部MRI检查显示双侧岛叶、海马回、双侧基底节区及半卵圆区广泛病灶;糖皮质激素治疗有效。根据其非侵蚀性、血清阴性多关节炎;眼部炎症(结膜炎、巩膜炎);耳蜗或前庭损害所致耳鸣、听力下降,耳郭软骨炎;以及激素治疗有效等表现,诊断首先应考虑复发性多软骨炎,但该病累及中枢神经系统者鲜见,国外已有相关报道,考虑可能与软骨蛋白多糖发生反应的Ⅱ型胶原蛋白抗体与神经系统(神经内膜、脑内小动脉中层及内膜)发生交叉反应有关。
诊治经过:根据入院后各项检查结果,临床诊断“颅内多发病灶”待查。患者入院后予抗病毒及营养神经治疗,以及糖皮质激素甲泼尼龙1g/d静脉冲击治疗3天后改为泼尼松60mg/d口服,治疗2周后减至50mg/d口服同时辅助环磷酰胺0.2g静脉注射,隔天一次。经治疗后患者症状明显改善、病情稳定。出院时诊断:复发性中枢神经系统炎性病变,系统性血管炎可能;耳郭软骨炎,复发性多软骨炎可能。建议继续口服泼尼松、环磷酰胺治疗。
最终耳郭组织病理报告为纤维、脂肪及软骨组织慢性炎性反应,支持以淋巴细胞浸润为主的炎性反应表现(图3),符合复发性多软骨炎的病理学特点。
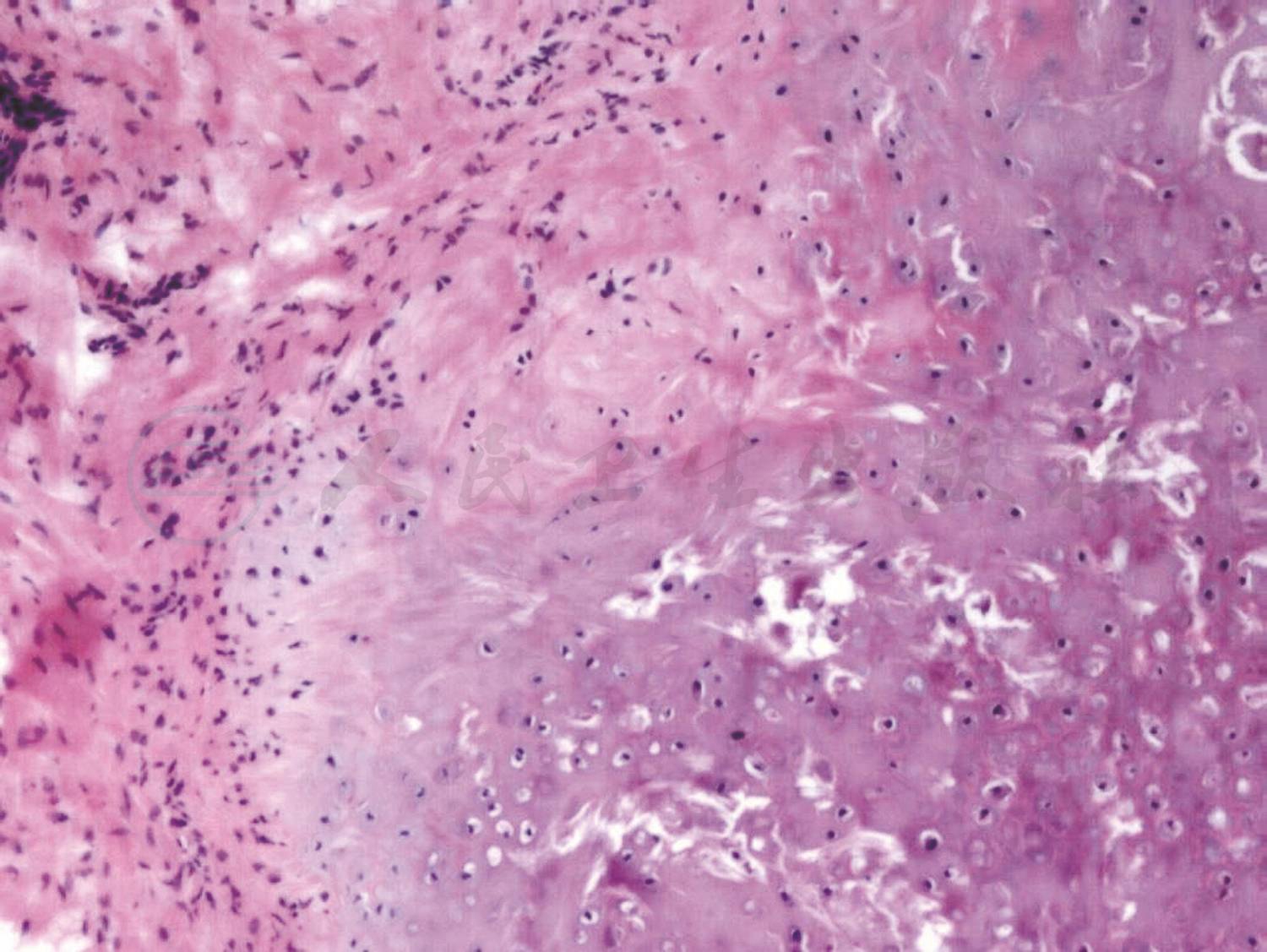
图3 光学显微镜所见
以淋巴细胞浸润为主的纤维、脂肪及软骨组织慢性炎性反应HE染色,×40
复发性多软骨炎(relapsing polychondritis,RP)
复发性多软骨炎为原因不明的由免疫介导的软骨炎性疾病,可以累及全身各个部位的透明软骨,最常见的是耳部的透明软骨炎。该病自第一次报道至今已有80多年。目前尚无确切的流行病学资料,但它可发生于所有种族及年龄组,且无性别差异性,估计发病率约占总人口的3/10万,发病年龄多为40~60岁,但亦有一些儿童发病的报道。虽然可能有遗传学因素参与了复发性多软骨炎的发生与发展过程,但目前仍未将其归于家族性遗传性疾病。
复发性多软骨炎的发病机制尚不清楚,推测其发病机制与体液免疫的参与有关。例如,软骨基质受到外伤、炎症、过敏等因素的影响使其抗原性暴露,从而引起一系列炎性反应,导致机体对局部软骨或有共同基质成分的组织产生免疫反应,包括巩膜、葡萄膜、玻璃体、视神经内膜及束膜、主动脉中层和内层的结缔组织、心瓣膜、心肌肌纤维膜、气管黏膜下基底膜、关节滑膜和肾小球及肾小管基底膜等。神经系统损害可能是对软骨蛋白多糖发生反应的抗Ⅱ型胶原抗体与神经系统(如神经内膜、脑内小动脉中层及内膜等)发生交叉反应所导致的相应病变。
复发性多软骨炎在严重程度和持续时间上呈多样性,以耳部损害为最常见的临床特征,同时可伴有其他具有透明软骨结构的器官受累,例如肋软骨、眼、呼吸道。①耳炎:最常见的特征,可以单侧或双侧炎症持续数天或数周。呈急性或亚急性发病,累及耳郭时表现为弥漫性红紫,亦可侵及外耳道影响听力。除此之外,约有30%耳部受累的复发性多软骨炎患者还可出现听力受损、耳鸣、由内耳炎症导致的眩晕等较为少见的临床表现,此与软骨炎所引起的咽鼓管结构破坏、内耳淋巴水肿或感音性耳聋有关。②关节炎:关节受累是仅低于耳炎的又一常见症状,其中70%以上的患者最终导致关节症状。单纯复发性多软骨炎患者的关节炎性反应呈现间断、游走性、非对称性、血清反应阴性且常为非侵蚀性的临床表现,典型患者手、足X线检查可见关节间隙变小和/或骨质减少,胸、锁关节,肋软骨,胸骨柄关节为常见受累部位。③眼炎:眼炎发生率占复发性多软骨炎患者的20%~60%。典型表现为巩膜外层炎、巩膜炎、溃疡性角膜炎、眼葡萄膜炎和突眼。④鼻软骨炎:与鼻软骨炎相伴发的症状有鼻外表皮肤变硬、鼻液外溢,以及鼻出血等。慢性炎症和软骨破坏可导致特征性的“鞍鼻”畸形。⑤神经系统炎性反应:中枢神经系统和周围神经系统受累约占复发性多软骨炎患者的3%,极为少见,但有些患者仅有神经系统受累表现。主要累及第2和第6~8对脑神经,也可以有痴呆、偏瘫、癫痫、脊髓炎、周围神经病、小脑功能不良、边缘性脑炎等。组织病理学检查提示受累血管被淋巴细胞、单核细胞、巨噬细胞侵润;但中枢神经系统大血管炎十分罕见,可导致动脉瘤形成或颈内动脉血栓,临床上类似脑血管意外。还有部分复发性多软骨炎患者在无任何临床症状出现时其头部MRI即已显示异常信号,为灰质和白质多发异常高信号,与血管炎的影像一致;故推测其病理改变可能是局灶性血管炎所致。脑脊液检查对复发性多软骨炎伴发神经系统损害者十分重要。主要表现为以淋巴细胞浸润为主的炎性反应,蛋白定量轻度升高,葡萄糖可无异常。对一些存在免疫缺陷的患者,应高度怀疑中枢神经系统感染,而脑脊液单纯疱疹病毒、真菌、隐球菌、结核等病原学检查有助于鉴别这些疾病。当与中枢神经系统感染进行鉴别诊断时,尤其是单纯疱疹病毒性脑炎,可于糖皮质激素冲击治疗前经验性应用阿昔洛韦或抗生素治疗。另外,复发性多软骨炎还同时伴有呼吸道、心脏、消化管、肾脏、皮肤等多器官损害的表现,并与其他疾病并存,例如约37%的复发性多软骨炎患者合并血液系统疾病、结缔组织病、血管炎、皮肤疾病或其他自身免疫性疾病。
目前针对复发性多软骨炎尚无特异性的实验室诊断指标,但一些炎性指标有助于诊断。在结合临床的基础上,血白细胞计数、血小板计数、红细胞沉降率和血清C反应蛋白水平的升高,均可作为参考依据,但复发性多软骨炎不存在特异性抗体。另外,还有一些免疫学检测指标提示其可能性,如抗中性粒细胞胞质抗体(ANCA)阳性率升高,类风湿因子、抗心磷脂抗体阳性,部分患者抗核抗体(ANA)亦可呈阳性,少数患者血清梅毒反应呈假阳性。而且,在部分复发性多软骨炎患者的血液中可出现抗Ⅱ型胶原抗体和抗软骨细胞抗体阳性,这些抗体靶向对抗自身或变性Ⅱ型胶原及蛋白聚糖,后两者通常广泛存在于破坏的软骨结构中,故有助于复发性多软骨炎的诊断,但均缺乏敏感性和特异性。
目前参考的标准,为1976年McAdam提出的诊断标准:①双耳软骨炎;②非侵蚀性多关节炎;③鼻软骨炎;④眼炎,包括结膜炎、角膜炎、巩膜炎、表层巩膜炎及葡萄膜炎等;⑤喉和/或气管软骨炎;⑥耳蜗和/或前庭受损,表现为听力丧失、耳鸣和眩晕。符合以下其中1项者即可诊断为复发性多软骨炎:①具有上述标准中3项或3项以上者;②具备其中1项并经组织病理活检证实者;③有两处或更多处不同解剖部位的软骨炎,对糖皮质激素或免疫抑制剂治疗有效者。如临床表现明显,并非每例患者均需行软骨活检。总之,复发性多软骨炎的诊断需要临床表现、实验室检查、影像学表现,必要时行受累组织活检等项检查,综合分析以明确诊断。
目前对复发性多软骨炎的治疗决策大部分基于临床经验和病例报告,根据疾病活动性和严重程度采取不同的药物治疗。对于仅有局灶性病变活动而无器官受累如仅耳、鼻软骨炎或周围性关节炎患者可给予非甾体抗炎药(NASID)治疗,但需密切观察其病情变化。当病情加重并出现器官受累时,如喉、支气管、心血管、肾、眼或神经系统损害者则应予以进一步治疗,应用糖皮质激素以抑制急性发作,减少复发频率及严重程度,初始剂量为0.50~1.00mg/(kg·d),分次或晨起一次顿服;对急性重症患者,可酌情增加糖皮质激素剂量甚至施行甲泼尼龙冲击疗法;待临床症状好转后,可逐渐减量以最小剂量维持1~2年或更长时间。亦可选择免疫抑制药以减少糖皮质激素长期应用,但其临床疗效尚无确切的循证医学证据,可能的药物不良作用和获益也在长期随访中进一步评估。常用的免疫抑制药包括环磷酰胺、硫唑嘌呤、环孢素、甲氨蝶呤,其中环磷酰胺为首选药物。
[1]Kent PD,Michet CJ,Luthra HS.Relapsing polychondritis.Curr Opin Rheumatol,2004,16(1):56-61.
[2]Yang SM,Chou CT.Relapsing polychondritis with encephalitis. Journal of Clinical Rheumatology,2004,10 (2):83-85.
[3]Foidrat JM,Abe S,Martin GM,et al.Antibodies to type Ⅱ collagen in relapsing polychondritis.N Engl J Med,1978,299(22):1203-1207.
[4]Isaak BL,Liesegang TJ,Michet CJ.Ocular and systemic findings in relapsing polychondritis.Ophthalmology,1986,93(5):681-689.
[5]Zeuner M,Straub RH,Rauh G,et al.Relapsing polychondritis:clinical and immunogenetic analysis of 62 patients.J Rheumatol,1997,24(1):96-101.
[6]Erten-Lyons D,Oken B,Woltjer R L,et al.Relapsing polychondritis:an uncommon cause of dementia.J Neurol Neurosurg Psychiatry,2008,79(5):609-610.
[7]Sundaram MB,Rajput AH. Nervous system complications of relapsing polychondritis. Neurology,1983,33 (4):513-515.
[8]Lahmer T,Treiber M,Werder AV,et al.Relapsing polychondritis:An autoimmune disease with many faces.Autoimmunity Reviews,2010,9(8):540-546.
[9]中华医学会风湿病学分会.复发性多软骨炎诊治指南(草案)中华风湿病学杂志,2004,8(4):251-253.
[10]乔雷,彭斌,关鸿志,等.复发性多软骨炎相关的中枢神经系统损害三例.中华医学杂志,2017,97(5):392-394.
[11]乔雷,李凌,王建明,等.反复关节疼痛眼痛耳廓红肿伴发热记忆力减退行为异常.中国现代神经疾病杂志,2010,10 (6):683-686.









