


 收藏
收藏 已收藏
已收藏患者女性,53岁。因“复发性双下肢麻木无力伴小便障碍4年”于2016年9月2日入院。
现病史
2012年10月12日患者无明显诱因出现双下肢无力、麻木,伴胸部束带感和后背部麻木、疼痛,热敷后自觉好转,未影响日常生活和活动。次日清晨出现左下肢无力加重,骑自行车时自觉左脚感觉不到踏板,当晚出现脐部以下麻木、感觉减退,双下肢行走拖步,蹲起不能,伴小便潴留;发病期间无发热、头痛、恶心、呕吐等,无视力障碍,无口眼歪斜、面部麻木、吞咽困难和言语障碍等,外院“活血化瘀”治疗(具体方案不详)无效,病情持续进展,2天后胸部以下皮肤无汗,麻木感上升至胸部以上,出现呼吸困难,声音低沉,饮水呛咳,不能进食,双上肢力弱、双下肢活动不能、小便障碍无缓解,当地医院行全脊椎MRI检查示C4~T5长节段脊髓长T1、长T2信号,伴脊髓肿胀,考虑脊髓炎,多发性硬化不除外。予激素治疗(具体方案不详)后症状逐渐好转,遗留有双下肢远端麻木感、行走不稳、小便控制略差。3年前(2013年7月5日)再次出现左下肢麻木、无力,不能行走,上肢持物费力,伴右眼视力减退、后背部疼痛、小便失禁,予甲泼尼龙冲击治疗3天后序贯甲泼尼龙100mg/d口服,下肢症状逐渐缓解,但视力未见明显好转。患者分别于2014年5月和2015年3月再次出现上述症状,激素治疗(具体方案不详)均有缓解;此后患者遗留有双下肢麻木无力、行走不稳、小便控制不佳及间断性便秘。自觉每次发作前诱发与缓解因素不明确。6个月前停用激素,症状较前进展。1个月前出现右眼水平方向复视,为求进一步诊断与治疗,就诊于我院。患者自发病以来,无眼干、口干、皮疹、雷诺现象、光过敏、关节肿痛,精神、睡眠、饮食尚可,大便次数减少,体重下降约5kg,曾因反复应用激素体重明显波动。
既往史
患者30年前反复出现双眼葡萄膜炎,继发左眼白内障致失明;6年前罹患甲状腺功能减退症,予甲状腺素替代治疗,1年前自行停药;3年前突发右眼视物不清,临床诊断为右眼白内障、瞳孔后粘连,激素治疗效果不佳,予右侧瞳孔成形术+白内障超声乳化术+晶状体植入术后视力略恢复;2年前外伤致脊柱压缩性骨折,予补钙治疗。
个人史
无特殊。
家族史
无特殊。
入院查体
体温36.7℃,脉搏72次/min,呼吸18次/min,血压90/51mmHg,经皮动脉血氧饱和度(SpO2)99%;轻度消瘦,胸椎轻度后凸畸形,皮肤偏黑,全身皮肤无皮疹,口腔黏膜无溃疡,无脱发,关节无肿胀和压痛,心肺腹部检查无异常。神经系统查体:神志清楚,语言流利,高级智能粗测正常;右眼视力0.5、视野正常,左眼失明、眼球萎缩凹陷不可查,右侧瞳孔不规则,直径3.00~3.50mm,直接对光反射迟钝,各向眼动充分,无眼震、复视;全身肌肉欠饱满,右上肢肌力4-级、左上肢5级、右下肢3级、左下肢4级,右下肢肌张力增高,余肢体肌张力正常;双侧指鼻试验和快复轮替动作稳准,左侧跟-膝-胫试验稳准、右侧欠稳准,睁眼与闭眼Romberg征均阳性,痉挛步态;双上肢腱反射活跃,尤以右侧显著,双下肢腱反射亢进,腹壁反射消失;双侧髌阵挛和踝阵挛阴性;双侧Babinski征和Chaddock征阳性;右侧面部针刺觉略减退,T4水平以下针刺觉、音叉振动觉和轻触觉均减退,T12以下音叉振动觉消失;脑膜刺激征阴性。
辅助检查
实验室检查血、尿、粪便常规、血液生化、凝血功能试验和感染四项均正常;红细胞沉降率(ESR)38mm/h,血IgG 24.16g/L(参考值范围7~17g/L),补体C3 0.62g/L(参考值范围0.73~1.46g/L)和C4 0.08g/L(参考值范围0.1~0.4g/L),类风湿因子(RF)158.20IU/ml(参考值范围0~20IU/ml),血清免疫固定电泳阴性;抗核抗体(ANA)阳性1:640,抗中性粒细胞胞质抗体(ANCA)、抗磷脂抗体谱均阴性,抗dsDNA-IF 1:10(<1:5)、抗dsDNA-ELISA 296IU/ml(<100IU/ml);肿瘤标志物筛查均呈阴性;血清同型半胱氨酸于正常值范围;血清铁(FeS)8.243µmol/L,转铁蛋白(TRF)18.06µmol/L,总铁结合力(TIBC)41.35µmol/L,转铁蛋白饱和度(TS)16.70%。腰椎穿刺脑脊液外观无色清亮,常规、生化和乳酸均于正常值范围,细胞学形态正常,髓鞘碱性蛋白(MBP)0.01nmol/L(<0.55),细菌涂片阴性,癌胚抗原(CEA)、甲胎蛋白(AFP)阴性,快速血浆反应素试验(RPR)阴性,抗水通道蛋白4(AQP4)抗体阳性1:100。胸部CT检查未见明显异常。头部MRI未见明显异常。脊椎MRI显示C7~T7长节段脊髓条状、斑点状长T2信号,T7~8椎体压缩性骨折(图1);腰椎退行性变,L4-5椎间盘内异常信号,L5~S1椎间盘膨出。唇腺组织活检提示淋巴细胞灶。
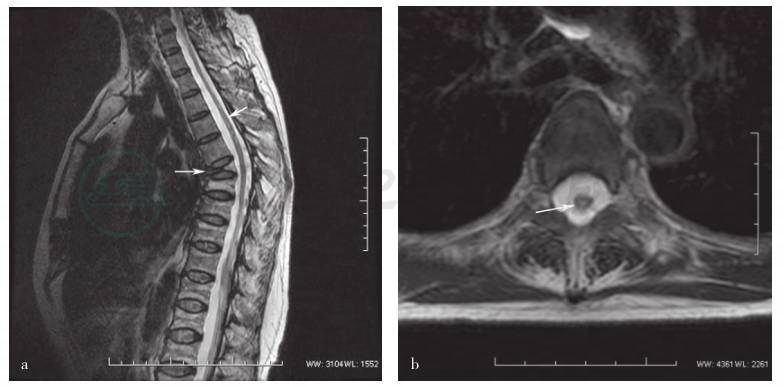
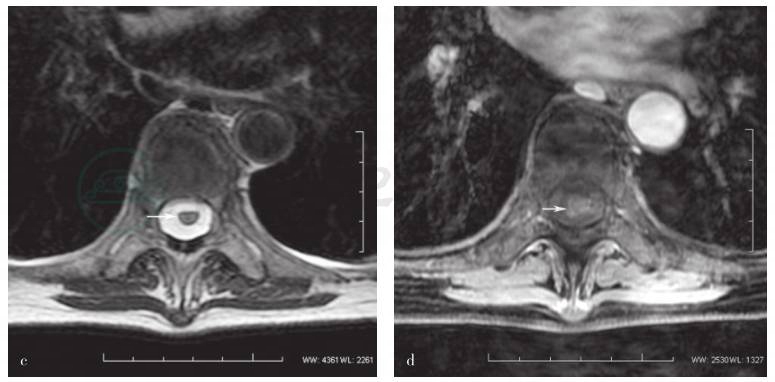
图1 患者胸椎MRI检查
a.矢状位T2WI显示C7~T7长节段脊髓内连续高信号(箭头所示),T7~10椎体压缩性骨折,尤以T7显著(箭头所示);b.横断面T2WI显示T6水平髓内偏右线样高信号(箭头所示);c.横断面T2WI显示、T8水平髓内斑片状高信号(箭头所示);d. T8水平病灶横断面T2WI钆强化效应不明显(箭头所示)
诊断与治疗过程
入院后予B族维生素(维生素B1 10mg,3次/d口服,甲钴胺0.5mg,3次/d口服)、巴氯芬(5mg,2次/d口服)治疗。临床诊断为“视神经脊髓炎谱系疾病,系统性红斑狼疮,继发性干燥综合征”。经内分泌科和骨科会诊后予甲状腺激素替代治疗(左甲状腺素钠 50µg,1次/d)和强化补钙治疗(碳酸钙500mg,1次/d口服,骨化三醇0.2µg,2次/d口服,阿仑膦酸钠维D3 70mg,1次/周口服),同时建议佩戴支具。自2016年9月13日起予甲泼尼龙500mg/d静脉冲击治疗3天,序贯泼尼松50mg/d口服,每2周减量5mg/d;同时增加硫唑嘌呤50mg,1次/d口服,长期维持;2个月后随访患者行走不稳较前改善,余无明显变化。
神经科主治医师
患者中年女性,急性发病,复发缓解病程4年;临床主要表现为反复肢体麻木、无力,严重时饮水呛咳、吞咽困难,激素治疗有效,每次发作后遗留部分后遗症。既往葡萄膜炎30年,左眼失明、右眼视力下降;体格检查双下肢肌力下降,腱反射亢进,病理征阳性,右侧跟-膝-胫试验欠稳准,睁眼与闭眼Romberg征均阳性,T4平面以下针刺觉、音叉振动觉和轻触觉减退,行走不稳,痉挛步态。定位诊断:①左眼失明、凹陷,右眼视力下降、视野粗测正常,定位于双侧视交叉前视神经,可能合并眼球内病变。②双下肢肌力下降,尤以右侧显著,双下肢肌张力略高、腱反射亢进,双侧病理征阳性,提示双侧锥体束受累,T4平面以下针刺觉、音叉振动觉和轻触觉减退,尤以左侧显著,提示脊髓丘脑侧束与后索病变,综合考虑脊髓T4平面以上横贯性病变,结合胸椎MRI,定位于T4水平以上脊髓。③走路不稳,睁眼与闭眼Romberg征均阳性,考虑脊髓小脑束可能受累。定性诊断:首先考虑炎性脱髓鞘病变,①视神经脊髓炎谱系疾病,患者中年女性,脊髓炎相关临床症状,脊椎MRI显示脊髓长节段受累,病程反复,激素治疗有效;此外,突发视力减退,视神经受累。视神经脊髓炎谱系疾病核心症状包括视神经炎、急性脊髓炎、最后区综合征、脑干综合征、发作性睡眠、大脑综合征伴特异性MRI征象,血清和脑脊液特异性抗体抗AQP4-IgG阳性。该例患者临床和影像学表现均提示视神经脊髓炎谱系疾病,完善血清和脑脊液抗AQP4-IgG检查、头部和眼眶MRI增强扫描以明确诊断。②Vogt-小柳-原田综合征(VKH),系多系统受累的自身免疫性疾病,可能与黑色素细胞抗原相关。好发于中青年女性,临床主要表现为双眼同时发病的急性葡萄膜炎,可伴听力异常、皮肤受累(毛发变白、白癜风、脱毛),累及中枢神经系统可以表现为脑膜炎、脑神经麻痹、横贯性脊髓炎等,脑脊液淋巴细胞计数增多,激素治疗有效。该例患者中年女性,发病急骤,既往有双眼葡萄膜炎和脊髓炎,激素治疗有效,可疑Vogt-小柳-原田综合征,须经眼科会诊以排除诊断。③系统性结缔组织病继发中枢神经系统损害,患者有眼部受累表现,且对激素治疗敏感。结缔组织病,如系统性红斑狼疮、白塞病和类风湿性关节炎等,常合并视神经脊髓炎谱系疾病,其中,白塞病常以双侧全葡萄膜炎为主要表现。该例患者无眼干、口干、光敏感、脱发和关节肿痛等免疫系统疾病症状,须完善感染相关指标以及抗核抗体、抗双链DNA抗体(dsDNA)、抗中性粒细胞胞质抗体等免疫指标以明确诊断。
神经科教授
患者中年女性,急性发病,病程反复,以反复发作的四肢麻木、无力伴吞咽困难、小便障碍为主要表现,伴有视神经损害,激素治疗有效;体格检查可见双侧视力损害,存在感觉平面,四肢肌力不同程度下降,腱反射亢进,共济失调;脊椎MRI显示脊髓长节段长T2信号,根据临床和影像学表现,拟诊炎性脱髓鞘疾病,结合视神经损害,根据国际视神经脊髓炎诊断小组(IPND)更新的2015年视神经脊髓炎谱系疾病诊断标准,考虑视神经脊髓炎谱系疾病可能性大。入院后完善血清和脑脊液抗AQP4-IgG检查,以及头部和眼眶MRI检查等,最终明确诊断。鉴别诊断方面:①多发性硬化(MS),患者女性,病程反复,激素治疗有效,有脊髓受累表现,应警惕多发性硬化,但脊椎MRI提示脊髓长节段连续病变,病变位于脊髓中央,头部MRI未见明显异常,故不支持诊断。②结节病,可以引起双眼葡萄膜炎和中枢神经系统病变,但多伴皮损和多系统结节表现。该例患者临床表现以中枢神经系统损害为主,故不支持诊断。③代谢和感染相关脊髓病变,亚急性脊髓联合变性、铜缺乏相关脊髓病均可出现脊髓长节段损害,但多为慢性发病且有全身系统性表现,血清叶酸和维生素B12水平可资鉴别。感染相关脊髓病如人类嗜T细胞病毒Ⅰ型(HTLV-1)、神经梅毒等,慢性发病,病程较长,进行性加重,脑脊液呈炎症反应表现且相关病毒或抗体阳性,须完善腰椎穿刺脑脊液检查以排除。④肿瘤或副肿瘤性病变,淋巴瘤进展缓慢,激素治疗有一定疗效。该例患者病程较长,肿瘤标志物筛查、脑脊液细胞学形态和脊椎MRI增强扫描均不支持诊断。治疗方面,大剂量激素滴注静脉冲击和序贯口服治疗联合免疫抑制剂治疗的同时,辅以B族维生素营养神经、巴氯芬降低肌张力、奥美拉唑抑制胃酸和保护胃黏膜、碳酸钙和氯化钾维持内环境稳定。该例患者视力较差,行动不便,应注意跌倒、摔伤等意外风险。
1.视神经脊髓炎谱系疾病(neuromyelitis optica spectrum disorder,NMOSD)
2.系统性红斑狼疮(systemic lupus erythematosus,SLE)
3.继发性干燥综合征(secondary Sjögren syndrome,SSS)
视神经脊髓炎(neuromyelitis optica,NMO)是免疫介导的严重特发性中枢神经系统炎性脱髓鞘性疾病,主要累及视神经和脊髓,发病机制与抗AQP4-IgG有关。1894年,Devic率先提出该病并认为是多发性硬化的一种表现为视神经炎和脊髓炎且单相病程的特殊亚型。20世纪以后,人们逐渐认识到大多数视神经脊髓炎患者呈复发病程,且伴特异性影像学和脑脊液淋巴细胞计数改变。2004年,Lennon等在视神经脊髓炎患者血清中检出高特异性NMO-IgG,从而确定该病是不同于多发性硬化的独立疾病。NMO-IgG于2006年首次纳入视神经脊髓炎的诊断标准中,并于2007年提出“视神经脊髓炎谱系疾病”的概念,包括视神经脊髓炎、长节段横贯性脊髓炎(longitudinally extensive transverse myelitis,LETM)和视神经炎(optic neuritis,ON),以及亚洲视神经脊髓型多发性硬化、伴系统性免疫性疾病的长节段横贯性脊髓炎和/或视神经炎、伴典型视神经脊髓炎颅内表现(如下丘脑、胼胝体、脑室旁或脑干病变)的长节段横贯性脊髓炎和/或视神经炎等。鉴于视神经脊髓炎与视神经脊髓炎谱系疾病在临床表现、影像学特点、治疗和预后等方面并无显著差异,且多数NMO-IgG阳性视神经脊髓炎谱系疾病患者最后亦进展为视神经脊髓炎,国际视神经脊髓炎诊断小组会议将两者统一命名为视神经脊髓炎谱系疾病,并于2015年提出该病最新诊断标准。视神经脊髓炎谱系疾病的核心临床症状包括视神经炎、长节段横贯性脊髓炎、最后区综合征、急性脑干综合征、症状性睡眠发作或急性间脑综合征伴典型间脑MRI病灶、症状性大脑综合征伴典型的脑病变;特征性影像学表现为脊髓病灶延伸≥3个椎体节段和最后区、室管膜周围病变,T2WI呈高信号,T1WI增强扫描呈强化征象;实验室检查根据NMO-IgG水平分为NMO-IgG阳性组和NMO-IgG阴性组,分别提出相应诊断标准。鉴于近50%视神经脊髓炎谱系疾病患者合并其他抗体阳性,如抗核抗体、抗干燥综合征A型和B型抗体(SSA和SSB)、抗甲状腺激素抗体等,合并上述抗体阳性更支持视神经脊髓炎谱系疾病的诊断。本例患者临床反复发作,有明确急性长节段脊髓炎表现,可疑视神经炎病史,脑脊液NMO-IgG阳性,脊椎MRI显示长节段脊髓炎征象,视神经脊髓炎谱系疾病诊断明确。
多种自身免疫性疾病如系统性红斑狼疮、干燥综合征、桥本甲状腺炎、原发性抗磷脂抗体综合征和重症肌无力均可合并视神经脊髓炎谱系疾病,尤其是血清NMO-IgG阳性患者。抗核抗体阳性在视神经脊髓炎谱系疾病患者中较为常见。北京协和医院的早期研究显示,与NMO-IgG阴性患者相比,NMO-IgG阳性患者血清抗核抗体阳性率(39.8%对5.9%,p=0.006)和血清总自身抗体(ANA+SSA+SSB)阳性率(55.7%对23.5%,p=0.019)均增加,提示NMO-IgG阳性患者自身免疫反应可能较阴性患者更剧烈。表位扩散假说基于水通道蛋白(AQP)各亚型之间氨基酸序列具有较高比例(19%~52%)的同源性,可以部分解释视神经脊髓炎谱系疾病与系统性红斑狼疮、原发性干燥综合征等疾病共存于同一例患者的现象。系统性红斑狼疮的典型中枢神经系统症状为癫痫发作、抑郁症状和多发性单神经炎等,表现为脱髓鞘病变者少见(<2%)。两者的病理生理学机制相关性尚不清楚。1999年美国风湿病学会(ACR)SLE分类标准描述了SLE患者的神经精神症状。事实上,关于系统性红斑狼疮合并视神经脊髓炎谱系疾病患者,最初难以明确是单独疾病还是系统性红斑狼疮的神经精神表现。Govoni等认为,视神经脊髓炎是神经精神狼疮的一类表现;而国际视神经脊髓炎诊断小组则认为,视神经脊髓炎谱系疾病可以合并自身免疫性疾病如系统性红斑狼疮、干燥综合征或重症肌无力等,且更支持视神经脊髓炎谱系疾病的诊断。对于此类患者,其中枢神经系统症状与体征很可能源于视神经脊髓炎谱系疾病,而非系统性红斑狼疮或干燥综合征并发症,如血管炎累及中枢神经系统。系统性红斑狼疮或原发性干燥综合征诊断明确而视神经炎或脊髓炎症状缺如患者,血清NMO-IgG阴性间接支持该推论。Birnbaum等回顾分析22例系统性红斑狼疮合并脊髓炎患者的临床资料发现,合并灰质病变者更易出现SLE活动,而合并白质病变者更倾向于视神经脊髓炎的诊断。本例患者病程反复、病情进行性加重,按照”复发性脊髓炎”予激素治疗,效果尚可,但出现骨质疏松等药物不良反应,尽管缺乏自身免疫性疾病主诉和肯定的阳性体征,但若能注意到炎性指标异常、合并甲状腺疾病等特点,早期筛查免疫学指标,或可能早期明确诊断,指导更完备的治疗方案。
视神经脊髓炎谱系疾病合并系统性红斑狼疮在治疗方面尚未达成共识。目前认为,大剂量激素冲击治疗后序贯环磷酰胺(CTX)静脉滴注联合泼尼松口服优于单纯激素治疗,主要用于控制急性期症状。维持期采用免疫抑制剂如硫唑嘌呤、吗替麦考酚酯或甲氨蝶呤,可以有效降低复发率。对于病程反复的难治性患者,美国神经病学学会(AAN)推荐利妥昔单抗(抗-CD20单抗),必要时可考虑血浆置换疗法。
综上所述,本例患者有急性脊髓炎临床表现伴特异性脊髓长节段连续病变、脑脊液NMO-IgG阳性,根据2015年国际视神经脊髓炎诊断小组共识明确诊断为视神经脊髓炎谱系疾病。此外,血沉等指标升高,免疫学指标ANA和dsDNA阳性、补体C3和C4降低,唇线组织活检术提示干燥综合征,均符合2009年美国风湿病学会系统性红斑狼疮诊断标准,明确诊断为结缔组织病、系统性红斑狼疮、继发性干燥综合征。鉴于自身免疫性疾病与视神经脊髓炎谱系疾病并存的比例较高,对于临床拟诊视神经脊髓炎谱系疾病的患者,尤其是病程反复者,应警惕合并自身免疫性疾病的可能。无论是否有结缔组织病变的临床表现,均应完善相关检查,做到早筛查、早诊断和早治疗。
[1]Wingerchuk DM,Banwell B,Bennett JL,et al.International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology,2015,85(2):177-189.
[2]Jarius S,Wildemann B.The history of neuromyelitis optica.J Neuroinflammation,2013,10:8.
[3]Lennon VA,Wingerchuk DM,Kryzer TJ,et al.A serum autoantibody marker of neuromyelitis optica:distinction from multiple sclerosis.Lancet,364(9451):2106-2112.
[4]Wingerchuk DM,Lennon VA,Lucchinetti CF,et al.The spectrum of neuromyelitis optica. Lancet Neurol,2007,6(9):805-815.
[5]Wingerchuk DM,Weinshenker BG.The emerging relationship between neuromyelitis optica and systemic rheumatologic autoimmune disease.MultScler,2012,18(1):5-10.
[6]张遥,费允云,牛婧雯.合并结缔组织病的视神经脊髓炎谱系疾病回顾性研究.中华医学杂志,2014,94(39):3056-3061.
[7]The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes.Arthritis Rheum,1999,42(4):599-608.
[8]Saison J,Costedoat-Chalumeau N,Maucort-Boulch D,et al.Systemic lupus erythematosus-associated acute transverse myelitis:manifestations,treatments,outcomes,and prognostic factors in 20 patients. Lupus,2015,24 (1):74-81.
[9]付瀚辉,张江涛,柳青,等.肢体无力麻木伴大小便障碍4年.中国现代神经疾病杂志,2017,17(3):235-239.









