


 收藏
收藏 已收藏
已收藏患者男性,61岁。因“头痛伴复视3个月余”于2017年11月21日入院。
现病史
入院前3个月余(2017年8月)患者出现左侧头痛,日间表现为左侧额顶部过电样疼痛,数字分级评分法(numerical rating scale,NRS)得分 4~5分,持续无缓解,夜间疼痛症状加重,表现为左侧额顶部搏动性胀痛,NRS 7~8分,无法入睡,自行服用布洛芬或氨酚咖匹林片后,头痛短暂性缓解,无恶心、呕吐,无发热。外院最初诊断不详,予头孢菌素类抗生素治疗5天,症状未见改善,并逐渐出现眩晕、复视,遮盖左眼后眩晕、复视可缓解,左眼左右视不能、上下视尚可,进一步行头部CT检查,临床考虑“缺血性卒中可能”,予药物治疗1.5个月,无明显缓解。14天前无明显诱因出现左侧齿龈和面部感觉减退,双耳偶有轰鸣音;11天前出现饮水呛咳、吞咽困难、言语不清、鼻音重,症状进行性加重至无法正常饮食,仅能进流食;外院行头部MRI增强扫描(2017年11月11日)显示左侧颅底硬脑膜增厚伴强化征象,眼眶CT未见明显异常,未予特殊治疗;5天前出现间断性午后发热,伴盗汗、乏力,体温最高38.5℃,多发生于晚上7点至8点,持续1~2小时后体温自行恢复,无畏寒、寒战,无咳嗽、咳痰、咯血。为求进一步诊断与治疗,至我院就诊。
患者病前有受凉流涕史。自发病以来,精神可,睡眠较差,因饮水呛咳、吞咽困难而饮食欠佳,小便正常,便秘,体重下降7kg,否认口干、眼干、光过敏、皮疹、口腔溃疡、关节肿痛和雷诺现象等。
既往史
患者有慢性鼻窦炎史。否认高热惊厥史和颅脑创伤史,无特殊物品接触史。
个人史
吸烟史20余年,20支/d,已戒烟20年;不饮酒,无疫区居住史。
家族史
否认家族中类似疾病病史,否认家族遗传性疾病病史。
入院查体
体温36.2℃,脉搏110次/min,呼吸20次/min,血压120/80mmHg,经皮动脉血氧饱和度(SpO2)94%;神志清楚,对答切题,粗测高级智力正常;双侧眼底视盘边界清晰,余未见明显异常,左侧眼裂大于右侧(左侧睑裂1.4cm,右侧1cm),左眼瞬目反射减少、辐辏反射无法完成,左眼闭目力弱,示齿口角向右偏斜,语音低、鼻音重,悬雍垂右偏,右侧咽腭弓稍高,双侧咽反射减弱;颈软无抵抗,四肢肌容积和肌力正常、肌张力增高,共济运动和深浅感觉检查未见明显异常,可直线行走,腱反射亢进,病理征阴性,脑膜刺激征阴性,自主神经系统未见明显异常。
辅助检查
入院后完善辅助检查,血常规白细胞计数13.40×109/L,中性粒细胞比例87%;肝功能试验血清谷丙转氨酶282U/L,谷草转氨酶190U/L;尿沉渣24小时尿蛋白定量0.52g,红细胞潜血25个/μl,红细胞计数9.70个/μl;尿常规、粪便常规、肾功能试验、电解质和凝血功能均于正常值范围;感染相关检测,结核菌素纯蛋白衍生物(PPD)、血清结核感染T细胞斑点试验(T-SPOT.TB)、血清巨细胞病毒(CMV)/EB病毒(EBV)DNA、感染免疫检测四项均呈阴性,2次痰培养均呈阴性,包括细菌和真菌培养、奴卡菌涂片、抗酸染色、六胺银(PASM)染色、墨汁染色、结核分枝杆菌(TB)/非结核分枝杆菌(NTM)DNA均呈阴性;免疫相关检测,红细胞沉降率(ESR)>140mm/h、超敏C反应蛋白(hsCRP)158.95mg/L(0~3mg/L),血清抗中性粒细胞胞质抗体(ANCA)IgG型免疫荧光法(IFA)呈阳性(<1:10)、髓过氧化物酶型 ANCA(MPO-ANCA)137RU/ml(<20RU/ml),抗核抗体(ANA)谱、抗可提取性核抗原(ENA)抗体、类风湿性关节炎(RA)相关自身抗体谱均于正常值范围,血清IgG及其亚型(IgG1、IgG2、IgG3、IgG4)于正常值范围。腰椎穿刺脑脊液外观清亮、透明,压力58mmH2O,细胞总数28×106/L、白细胞计数22×106/L、单核细胞计数22×106/L,蛋白定量980mg/L、葡萄糖和氯化物于正常值范围;病原学筛查呈阴性,包括细菌涂片和培养、真菌涂片、奴卡菌涂片和培养、墨汁染色、抗酸染色、结核分枝杆菌/非结核分枝杆菌DNA测定、梅毒螺旋体明胶凝集试验(TPPA)和快速血浆反应素试验(RPR)、EB病毒衣壳抗原IgM(VCA IgM)、布鲁氏菌虎红试验、隐球菌抗原测定、抗莱姆病抗体、脑囊虫抗体均呈阴性;脑脊液细胞学检查白细胞计数1000/0.50ml、淋巴细胞比例0.90,呈淋巴细胞性炎症反应;寡克隆区带(OB)和副肿瘤相关抗体(抗Hu、Yo、Ri抗体等)于正常值范围。影像学检查:头部MRI增强扫描(2017年11月11日)显示左侧海绵窦、小脑幕和颅底硬脑膜增厚伴强化征象(图1),考虑炎症性病变可能性大。PET/CT显示右侧肺尖斑片影,代谢稍增高,双肺支气管血管束走行区多发代谢增高,考虑炎症性病变。胸部增强CT和高分辨率CT(HRCT)显示双肺支气管血管束增粗,支气管壁多发増厚,双肺散在索条影,右肺下叶钙化点,双侧肺门和纵隔多发小淋巴结,部分钙化。眼眶CT未见明显异常。
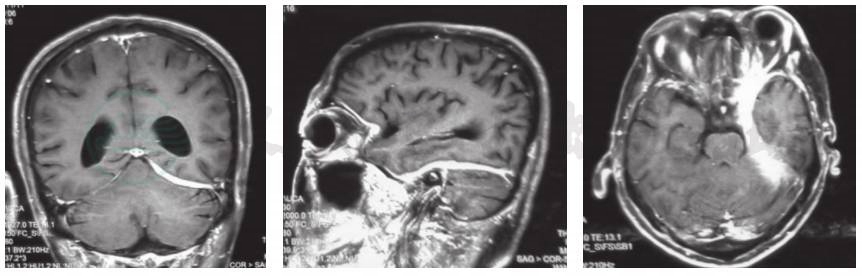
图1 患者治疗前头部MRI检查
a.冠状位增强T1WI显示左侧小脑幕和颅底硬脑膜增厚伴强化征象;b.矢状位增强T1WI显示硬脑膜广泛性增厚伴线性强化征象;c.横断面增强T1WI显示左侧海绵窦、颞区硬脑膜增厚伴强化征象
1.神经科主治医师
患者老年男性,急性发病,病程3个月余;临床以左侧额顶部胀痛、复视、左眼外展受限为主要表现,进展较快,出现饮水呛咳、吞咽困难,伴发热、盗汗、体重下降;既往有鼻炎病史;体格检查可见左侧眼裂偏大,双眼左视略慢,左眼瞬目反射减少、闭目反射偏弱,示齿口角右偏,语音低、鼻音重,悬雍垂右偏;头部MRI增强提示左侧颅底硬脑膜增厚伴强化征象。
2.定位诊断
患者临床症状与体征提示左侧多支脑神经受累,包括动眼神经、三叉神经、动眼神经、面神经、舌咽神经及迷走神经等,以颅中窝、颅后窝受累为主,包括海绵窦区等。头部MRI增强提示左侧硬脑膜受累,临床表现、体格检查与影像学相符,定位于左侧硬脑膜及脑神经。
3.定性诊断
患者急性发病,有头痛等刺激性症状,多支脑神经受累表现,结合头部MRI增强扫描,肥厚性硬脑膜炎诊断明确。病因方面包括:
(1)自身免疫性脑膜炎
①抗中性粒细胞胞质抗体相关脑膜炎,患者血清ANCA阳性,其中MPO-ANCA显著升高;免疫学指标升高;尿常规和尿沉渣检测、胸部增强CT和高分辨力CT提示肾脏和肺受累,抗中性粒细胞胞质抗体相关血管炎诊断明确。腰椎穿刺脑脊液检查提示非特异性炎症性改变,考虑肥厚性硬脑膜炎,继发于抗中性粒细胞胞质抗体相关血管炎可能性大,必要时可行硬脑膜组织活检术明确诊断。②IgG4相关硬化性硬脑膜炎,患者血清IgG及其亚类于正常值范围,为不支持点,但临床尚需进一步明确,硬脑膜活检有助于诊断。
(2)中枢神经系统感染
①结核性脑膜炎,患者有发热,伴盗汗、消瘦等全身症状;免疫学指标升高;PET/CT提示右肺尖斑片影,考虑全身炎症反应明显。而且,结核分枝杆菌感染易于颅底出现脑膜炎和硬脑膜炎,与患者病变位置一致,故不能排除结核分枝杆菌感染,但结核分枝杆菌感染常同时累及硬脑膜和软脑膜,多为双侧受累,而该例患者无软脑膜受累,且PPD试验和T-SPOT.TB试验、痰液和脑脊液抗酸染色、结核分枝杆菌/非结核分枝杆菌DNA测定均呈阴性,为不支持点。②其他,患者既往有鼻窦炎病史,此次有鼻窦炎发作,不能排除其他细菌、真菌感染,但血清、痰液和脑脊液细菌、真菌、病毒等病原学检测均呈阴性,为不支持点。
(3)肿瘤相关脑膜炎
患者老年男性,病程中消瘦明显,不能排除肿瘤,尤其是转移瘤,但肿瘤标志物均呈阴性,PET/CT未见明显异常,考虑恶性肿瘤可能性不大,必要时可行脑膜组织活检术以进一步排除。
4.神经科教授
患者以头痛为首发症状,病程中消瘦明显,一般状况较差;血清免疫学指标(红细胞沉降率、超敏C反应蛋白)明显升高;头部MRI增强扫描显示左侧颅底硬脑膜增厚伴强化征象;既往有鼻窦炎病史;结合临床表现、实验室和影像学检查,肥厚性硬脑膜炎诊断明确。病因方面考虑中枢神经系统感染或免疫性疾病所致。患者病程中的脑神经症状具有一定可复性,脑脊液细胞学以淋巴细胞反应为主,未见中性粒细胞,故考虑免疫因素介导,抗中性粒细胞胞质抗体相关血管炎可能性大。但患者病变主要累及单侧中颅窝底,范围较局限,有发热症状,既往有鼻窦炎病史,发病前有鼻窦炎发作,仍不能完全排除感染因素,且抗中性粒细胞胞质抗体相关血管炎易合并感染,考虑先予激素治疗,观察病情变化,警惕免疫抑制治疗后可能继发的感染或原有感染加重,若激素疗效不佳,待肝功能恢复正常后,在充分排除感染的基础上,谨慎加用免疫抑制剂。建议向患者及其家属充分告知病情,交代相关风险和获益,行硬脑膜组织活检术明确病变性质。
5.诊治经过
临床诊断为“抗中性粒细胞胞质抗体相关血管炎,继发性肥厚性硬脑膜炎”。为排除感染,明确病理性质后指导治疗,建议行硬脑膜组织活检术,患者及其家属顾虑相关风险拒绝。予泼尼松50mg/次、1次/d口服,每2周减量5mg,莫西沙星0.40g/次、1次/d口服抗感染,曲马多50mg/次、1次/晚口服镇痛,治疗10天后肝功能试验明显升高,考虑药物相关肝损伤可能,停用莫西沙星和曲马多,改为多烯磷脂酰胆碱(易善复)456mg/次、3次/d和甘草酸二铵(甘利欣)150mg/次、3次/d口服改善肝功能,体温恢复正常,症状明显好转。患者共住院28天,出院后1个月门诊随访,症状无反复,复查头部MRI增强显示增厚的硬脑膜变薄,强化征象减轻,病变范围缩小(图2)。
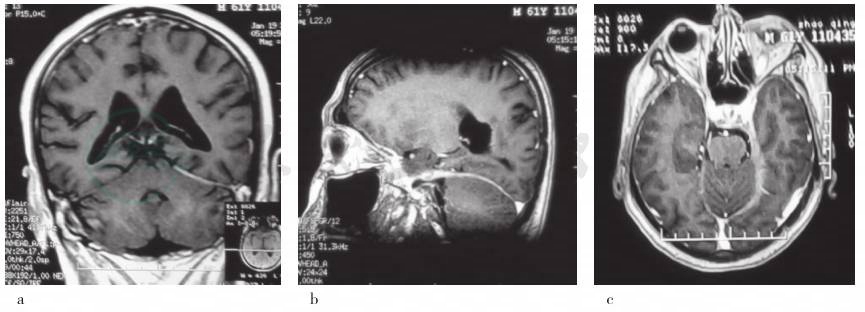
图2 治疗后复查头部MRI增强
a.冠状位增强T1WI显示左侧小脑幕病变范围缩小;b.矢状位增强T1WI显示增厚的硬脑膜变薄,病变范围缩小;c.横断面增强T1WI显示左侧海绵窦、颞区硬脑膜变薄,病变范围缩小
抗中性粒细胞胞质抗体相关血管炎(ANCA associated vasculitis,AAV)
继发性肥厚性硬脑膜炎(secondary hypertrophic pachymeningitis,SHP)
肥厚性硬脑膜炎(hypertrophic pachymeningitis,HP)是一种以硬脑膜肥厚和纤维化炎症反应过程为特征的少见疾病,由Charcot于1869年首次描述。近年来,国内外文献报道增多,Yonekawa等报告159例肥厚性硬脑膜炎患者,主要发生于颅底、小脑幕和大脑镰等部位,好发于成人,男性比例略高于女性,平均发病年龄约58岁,呈急性或亚急性发病,多为复发-缓解病程。研究显示,其临床特征主要是慢性头痛和多发性脑神经功能障碍,慢性剧烈头痛为最常见的首发症状,多数患者同时出现相应部位的脑神经损伤;肥厚性硬脑膜直接压迫邻近脑组织或影响静脉回流,也可以出现相应脑实质受累表现,以小脑性共济失调、癫痫发作常见;少数患者可以出现颅内静脉窦血栓形成、广泛性自主神经功能障碍、阻塞性脑积水等。
MRI对肥厚性硬脑膜炎有重要诊断价值,主要表现为硬脑膜局灶性或弥漫性增厚,可见结节状、线样病灶,静脉窦受累叶可以表现为结节状病灶;T1WI呈现与脑皮质相同的等或略低信号、T2WI呈低信号,增强扫描病灶可见明显强化。
肥厚性硬脑膜炎的发病机制尚不明确,根据病因可以分为特发性肥厚性硬脑膜炎(idiopathic hypertrophic pachymeningitis,IHP)和继发性肥厚性硬脑膜炎,常见的继发因素主要包括:①感染性疾病,如急慢性中耳炎、鼻窦炎,以及梅毒、结核分枝杆菌、隐球菌感染等。病原菌常通过颅骨骨折、中耳炎、鼻窦炎等疾病感染硬脑膜,可行硬脑膜组织活检术或细菌培养以明确诊断。然而硬脑膜组织活检细菌学呈阴性并不能完全排除感染的可能。Parney等报告1例肥厚性硬脑膜炎患者,硬脑膜组织活检术细菌学呈阴性,但结合其结核病疫区居住史、PPD试验强阳性等病史,予抗结核治疗后,肥厚性硬脑膜炎相关临床表现和影像学明显改善。②自身免疫性疾病,如IgG4相关疾病、抗中性粒细胞胞质抗体相关血管炎、风湿性关节炎、结节病等,故认为该病与机体免疫异常密切相关。③其他,如肿瘤引起的异常免疫反应(副肿瘤综合征)也可以导致肥厚性硬脑膜炎。对一些病因不明的肥厚性硬脑膜炎,排除继发性因素后,可以诊断为特发性肥厚性硬脑膜炎。肥厚性硬脑膜炎病理学呈慢性、非特异性炎症改变,纤维增生明显、慢性炎性细胞浸润,10%可见肉芽肿样改变;因其对激素治疗有效,故认为是一种自身免疫性疾病。
近年来,关于ANCA阳性肥厚性硬脑膜炎的报道越来越多,尤其来自日本的研究报道较多。肥厚性硬脑膜炎可以继发于抗中性粒细胞胞质抗体相关血管炎(AAV),且几乎仅继发于肉芽肿性多血管炎(GPA);临床表现以头痛、脑神经麻痹、副鼻窦炎为主,全身系统受累轻微;实验室检查抗髓过氧化物酶抗体(MPO-ANCA、P-ANCA)和抗人类中性蛋白酶3抗体(PR3-ANCA,C-ANCA)阳性率无明显差异;病理学显示硬脑膜肥厚,并可见肉芽肿型炎症反应、血管炎、多核巨细胞等;免疫治疗后硬脑膜病变明显改善。部分学者认为,抗中性粒细胞胞质抗体相关肥厚性硬脑膜炎可能是抗中性粒细胞胞质抗体相关血管炎,特别是肉芽肿性多血管炎的早期表现或特殊临床表现。
研究显示,部分ANCA阳性肥厚性硬脑膜炎患者临床主要表现为头痛、脑神经受累,全身症状轻微,几乎无血管炎相关临床证据;病情活动期,C-ANCA阴性、P-ANCA阳性,部分伴抗核抗体或类风湿因子阳性,与抗中性粒细胞胞质抗体相关血管炎继发肥厚性硬脑膜炎的临床特征不符,且肉芽肿性多血管炎的特异性抗体为C-ANCA或PR3-ANCA;而这部分患者病情活动期C-ANCA阴性、P-ANCA阳性,故不符合肉芽肿性多血管炎诊断标准;病程中无呼吸道、肾脏等多脏器受累证据,因此提出P-ANCA相关肥厚性硬脑膜炎的诊断,认为其可能是一种独立的疾病实体。
目前,国内外关于P-ANCA相关肥厚性硬脑膜炎的报道较少,根据文献总结其临床特征:①好发于50~70岁,无明显性别差异;②多无明显诱因,呈亚急性发病;③常见临床症状为头痛,其次为脑神经损害,亦可有其他症状,且全身症状轻微,部分患者无呼吸道和肾脏等多脏器受累证据;④病情活动期C-ANCA阴性、P-ANCA阳性,可伴抗核抗体或类风湿因子阳性;⑤血清炎症反应指标升高,如红细胞沉降率、超敏C反应蛋白明显升高且与病情进展高度相关;⑥激素联合免疫抑制剂治疗效果优于激素单药治疗。
[1]Shigeo R,Shigenori K.Idiopathic hypertrophic pachymeningitis.Neuropathology,2010,23 (4):335-344.
[2]Yonekawa T,Murai H,Utsuki S,et al.A nationwide survey of hypertrophic pachymeningitis in Japan.J Neurol Neurosurg Psychiatry,2014,85 (7):732-739.
[3]Mamelak AN,Kelly WM,Davis RL.Idiopathic hypertrophic cranial pachymeningitis:report of three cases.J Neurosurg,1993,79 (2):270-276.
[4]Nishioka H, Ito H,Haraoka J.Idiopathic hypertrophic cranial pachymeningitis of the cavernous sinus mimicking lymphocytic hypophysitis.Neurol Med Chir,1998,38 (6):377-382.
[5]Huang Y,Chen J,Gui L.A case of idiopathic hypertrophic pachymeningitis presenting with chronic headache and multiple cranial nerve palsies:A case report. Medicine,2017,96 (29):e7549.
[6]Parney IF,Johnson ES,Allen PB.“Idiopathic”cranial hypertrophic pachymeningitis responsive to antituberculous therapy:case report. Neurosurgery,1997,41 (4):965-971.
[7]Yokoseki A,Saji E,Arakawa M,et al.Hypertrophic pachymeningitis:significance of myeloperoxidase antineutrophil cytoplasmic antibody. Brain,137 (2):520-536.
[8]高彬洋,卢强,黄颜,等.头痛伴复视3月余.中国现代神经疾病杂志,2018,18 (4):293-296.









