


 收藏
收藏 已收藏
已收藏患者男性,42岁。因“进行性四肢麻木、无力4年”于2016年3月24日入院。
现病史
患者4年前(2012年初)无明显诱因出现全身乏力、剧烈运动耐力下降、易疲劳,症状无日间波动;此后,逐渐出现双侧手指、足趾麻木,伴双手握力下降,日常生活活动能力无明显降低,无肌肉疼痛、视物模糊等,未予重视。约3年前(2013年6月)出现双下肢麻木、无力,下山、上楼费力,有时蹲起困难,伴脚踏不实、行走不稳,走路时呈醉酒步态;此后,逐渐出现双手骨间肌萎缩,尤以鱼际明显,双足略下垂,穿衣、系纽扣笨拙,同时麻木感逐渐进展至手腕和脚踝。外院实验室检查血清肌酸激酶(CK)411U/L,餐后血糖14.80mmol/L,临床诊断为“糖尿病,糖尿病周围神经病变”,予以控制血糖、营养神经等治疗,血糖逐渐降至正常水平,但四肢麻木、无力症状仍持续加重,逐渐出现双侧足背屈不能、脚踏力减弱,行走时需用力将腿抬高以避免绊倒,蹲起不能,伴踩棉花感,行走不稳尤以夜间显著;双手精细活动逐渐不能完成,并出现小腿变细,无大小便障碍,无明显肉跳感。2015年底外院神经电生理学检查神经传导速度(NCV)显示上下肢周围运动神经和感觉神经均未引出波形,肌电图呈神经源性损害;影像学检查颈椎MRI未见明显异常,临床诊断为“糖尿病酮症酸中毒,糖尿病周围神经病变可能,肌萎缩侧索硬化症可能”,继续强化降血糖和营养神经治疗,临床症状无缓解,仍缓慢进展。约3个月前(2016年1月)开始出现双侧手指、足趾刺痛感,无肌肉疼痛或触(握)痛,无饮水呛咳、吞咽困难,无发汗异常。为求进一步诊断与治疗,至我院门诊就诊,门诊体格检查可见以四肢远端为主的运动和感觉异常,遂以“周围神经病,进行性神经性腓骨肌萎缩症待排除,糖尿病”收入院。患者自发病以来,精神尚可,睡眠良好,存在口干、多食、多饮和多尿,体重下降约10kg,无皮疹、雷诺现象、光过敏,不伴关节肿痛。
既往史、个人史及家族史
患者既往痛风病史16年,间断服药(具体方案不详),控制良好;生长发育良好,体育成绩佳,吸烟20年,平均20支/d,无饮酒,无特殊毒物接触史;父亲、胞姊可疑四肢远端肌萎缩、肢体偏细,胞兄身体健康,外甥和侄子自幼体质较弱、四肢远端纤细,日常生活和活动能力未见明显改变。
入院后体格检查
体温36℃,脉搏76次/min,呼吸19次/min,血压118/81mmHg(1mmHg=0.133kPa),经皮动脉血氧饱和度(SpO2)99%;体形消瘦,神志清楚,语言流利,对答切题,粗测高级智能正常,脑神经未见明显异常;四肢肌萎缩,尤以远端显著,呈“鹤腿”样,高足弓,无翼状肩胛;双上肢近端肌力5-级,肱三头肌肌力3级,双手并指、外展、对指力均较弱,呈“爪形手”,双下肢近端肌力4-级,双足背伸肌力2级,跖屈肌力4-级,肌张力均正常;跨阈步态,行走直线不能;四肢腱反射消失,腹壁反射未引出,病理反射未引出,双踝以下针刺觉减退,余肢体针刺觉、音叉振动觉、关节位置觉和复合觉未见明显异常;双侧指鼻试验、跟-膝-胫试验阴性,Romberg征阳性,脑膜刺激征阴性,自主神经系统检查未见明显异常。
入院后辅助检查
实验室检查血、尿、粪便常规、甲状腺功能试验、血清脂质、叶酸、红细胞沉降率、凝血功能试验和感染免疫检测四项均于正常值范围;血糖12.20mmol/L,糖化血红蛋白7.60%,血清尿酸454μmol/L,维生素B12(服用甲钴胺后)777pmol/L(80~675pmol/L);抗核抗体(ANA)谱(18项)、抗可提取性核抗原(ENA)抗体、抗中性粒细胞胞质抗体(ANCA)谱均于正常值范围,血清免疫固定电泳(IFE)阴性;血清肿瘤标志物筛查均呈阴性。腰椎穿刺脑脊液检查外观清亮透明,压力120mmH2O,常规于正常值范围,蛋白定量1.07g/L、葡萄糖6.50mmol/L,氯化物为正常值范围,墨汁染色、抗酸染色、真菌涂片、细菌涂片和隐球菌抗原均呈阴性,髓鞘碱性蛋白(MBP)为正常值范围,脑脊液IgG 101mg/L(10~40mg/L),寡克隆区带阳性、特异性寡克隆区带阳性;血清IgG为正常值范围,寡克隆区带阴性。血清和脑脊液抗Hu、Ri、Yo抗体,抗 CV2/CRMP5抗体,抗PNMA2(Ma2/Ta)抗体,抗双载蛋白(amphiphysin)抗体均呈阴性;血清和脑脊液抗莱姆病螺旋体抗体(IgG)、抗神经节苷脂抗体GM1(IgG+IgM)均呈阴性。影像学检查:肝、胆、胰、脾、双肾超声显示胆囊壁毛糙,胆囊结石;双肾、输尿管、膀胱、前列腺超声显示前列腺稍大;双侧颈动脉、椎动脉、锁骨下动脉彩色多普勒超声(CDUS)和胸部X线未见明显异常;双侧正中神经和尺神经超声显示横截面面积明显增大,提示神经增粗(图1)。神经电生理学检查:节段性运动神经传导未见传导阻滞(运动神经传导波幅降低,远端潜伏期延长,双侧桡神经运动传导速度减慢);神经传导速度(NCV)提示四肢周围神经源性损害,包括感觉纤维和运动纤维(运动神经传导速度均<38m/s);肌电图提示四肢、胸旁肌神经源性损害;四肢皮肤交感反应(SSR)未见明显异常。基因检测显示,周围神经髓鞘蛋白22(PMP22)外显子1_5重复突变(EX1_5 DUP),为杂合子。
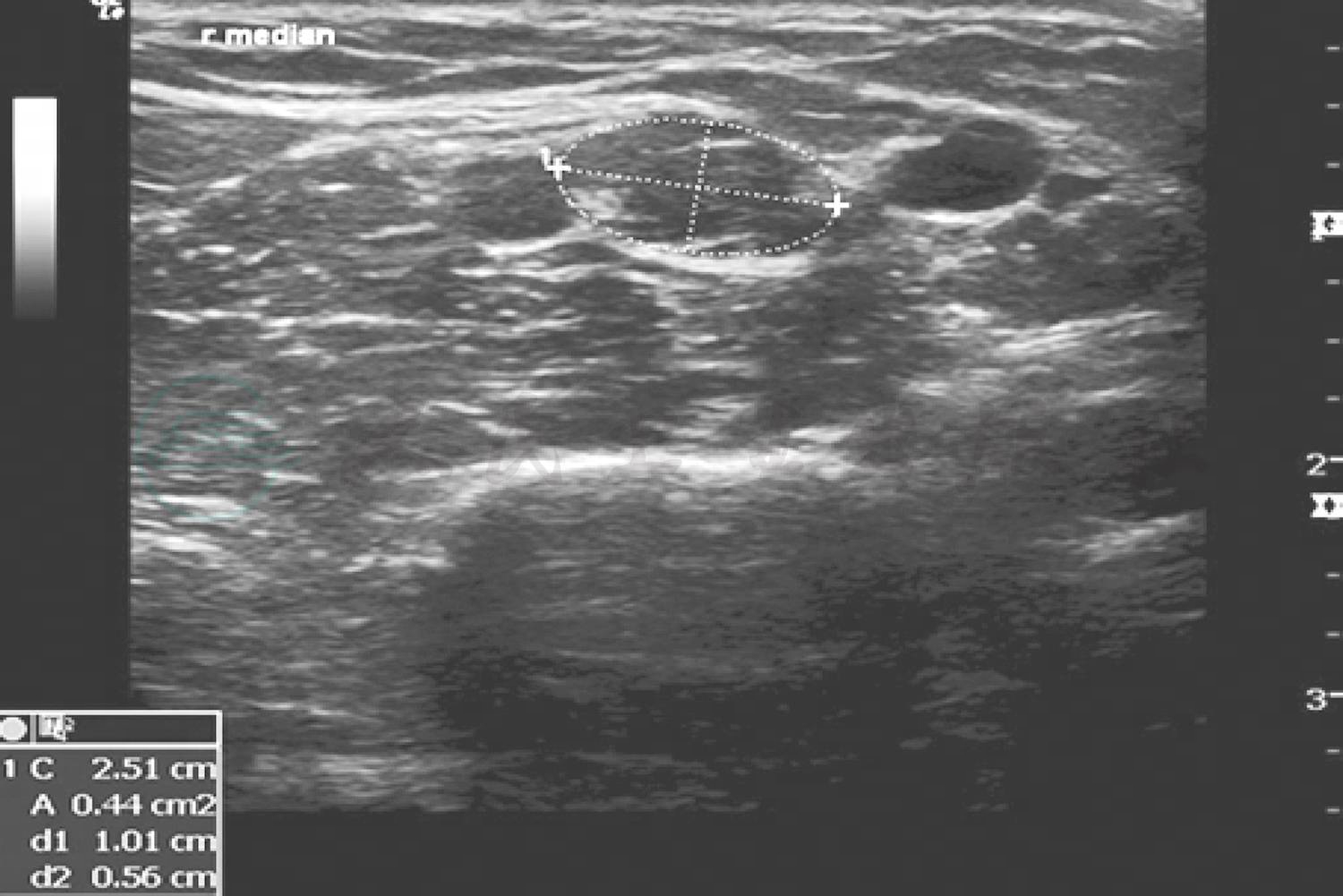
图1 患者右侧正中神经上臂段横断面超声
显示神经明显增粗[神经横截面积(CAS)0.44cm2(正常参考值≤0.10cm2)]
诊断与治疗经过
临床诊断“周围神经病变”明确,考虑遗传性周围神经病变可能性大,不排除糖尿病周围神经病,结合基因检测结果,最终诊断为遗传性运动感觉神经病(hereditary motor sensory neuropathy,HMSN),即腓骨肌萎缩症(Charcot-Marie-Tooth disease,CMT)。结合临床表现、神经电生理学检查和基因检测结果,诊断为腓骨肌萎缩症1A型。予维生素B1 10mg/次(3次/d)口服15日,维生素B12 500μg/d肌内注射7日后改为500μg/次(3次/d)口服8日,辅酶Q10 10μg/次(3次/d)口服15日,门冬胰岛素皮下注射控制并监测血糖。患者共住院15日,出院后持续在门诊进行康复训练并佩戴下肢矫形肢具,6个月后随访,运动功能有所好转,感觉症状无明显加重。
神经科主治医师
患者中年男性,隐匿起病,总病程4年;以四肢无力发病,逐渐出现四肢麻木、肌肉萎缩,尤以远端显著,疾病后期伴踩棉花感和步态改变;上述症状缓慢进行性加重无缓解;病程中无大小便障碍和自主神经功能障碍。既往有糖尿病和痛风病史。体格检查可见四肢肌无力、肌萎缩,尤以远端显著;四肢“手套-袜套”样针刺觉减退,尤以远端显著;Romberg征阳性;呈跨阈步态;双侧高弓足。定位诊断:感觉方面,对称性四肢麻木感自远端向近端进展,疾病后期出现手指、足趾刺痛感,并行走不稳、直线行走不能和踩棉花感,体格检查可见双踝以下针刺觉减退,呈末梢型、非传导束样分布,无小脑性共济失调表现和脊髓后索受累体征,定位周围神经;运动方面,对称性肌力减弱和肌萎缩,尤以远端显著,体格检查可见“鹤腿”,高弓足,四肢腱反射消失,病理征阴性,定位下运动神经元。结合肌电图,定位周围神经,感觉和运动纤维均受累。患者双侧桡运动神经传导速度检测显示远端潜伏期明显延长、传导速度减慢,提示髓鞘损害;多条神经(双侧正中神经和尺神经)运动传导波幅明显降低或消失,针极肌电图显示神经源性损害,提示继发性轴索损害。定性诊断:患者中年男性,隐匿起病,进行性加重,病史较长;周围性感觉和运动障碍表现突出,临床症状相对较轻而肌萎缩程度较重,可疑四肢肌萎缩家族史,基因检测显示PMP22外显子1_5重复突变,遗传性运动感觉性周围神经病诊断明确。
鉴别诊断应包括代谢性、免疫性和异常蛋白血症等相关周围神经病。
(1)代谢性周围神经病
①糖尿病周围神经病(diabetic peripheral neuropathy,DPN),以2型糖尿病为主,病程>10年者更易发生,临床表现为对称性肢体远端感觉运动神经病,小纤维损伤突出,主要以感觉障碍为主,自主神经系统亦受累,表现为大小便障碍、腹胀、心律异常、发汗异常等。糖尿病周围神经病具有长度依赖性,多由下肢起病,尤以远端显著,肌萎缩出现时间较晚,肌萎缩程度与肌力呈负相关。该例患者既往有2型糖尿病病史,血糖控制不佳,感觉和运动障碍自远端向近端进展,尤以远端显著,然而血糖控制良好后病情仍缓慢进展,肌萎缩明显,感觉障碍不突出,此为不支持点。②维生素B12和叶酸缺乏可以导致以后索和侧索受累为主的亚急性联合变性。然而该例患者既往无相关消化系统疾病或营养缺乏病史,体格检查显示本体感觉障碍不突出,无侧索受累,营养神经治疗无效,此为不支持点。③甲状腺功能异常致周围神经病,该例患者甲状腺功能试验和甲状腺超声均未见明显异常,故可排除诊断。
(2)免疫性周围神经病
如慢性炎性脱髓鞘性多发性神经病(chronic inflammatory demyelinating polyneuropathy,CIDP),该例患者慢性病程,感觉和运动系统均受累,神经传导速度检查提示周围神经髓鞘损害,脑脊液蛋白定量升高,需考虑慢性炎性脱髓鞘性多发性神经病。不支持点为弓形足,病程较长,节段性神经传导未见传导阻滞。慢性炎症性脱髓鞘性多发性神经病免疫治疗有效可资鉴别。此外,其他系统性自身免疫性疾病和副肿瘤综合征也可出现周围感觉和运动神经受累,但系统性自身免疫性疾病累及周围神经系统多表现为多发性单神经病。该例患者一般状况良好,临床无多系统受累症状,免疫指标和肿瘤标志物筛查均呈阴性,故不支持诊断。
(3)异常蛋白血症性周围神经病
可表现为周围神经病变,感觉和运动系统均受累,该例患者无其他系统受累表现,血清免疫固定电泳阴性,故不支持诊断。
(4)其他
如中毒性、感染性周围神经病,该例患者无明确毒物、神经毒性药物接触史,病程较长,无相关感染病史,可排除诊断。
神经科教授
患者中年男性,慢性进展性病程,以四肢远端感觉和运动障碍发病,肌萎缩突出,双侧对称,自主神经系统受累不明显,神经电生理学检查以脱髓鞘改变为主,周围性运动感觉神经病诊断明确。病因方面,应从获得性和遗传性方面考虑,前者包括特发性疾病、系统性疾病(如糖尿病、慢性感染)、自身免疫性疾病(如慢性炎性脱髓鞘性多发性神经病)、环境因素诱导和中毒性疾病等,其中,特发性周围神经病常发生于50岁以上人群,以感觉障碍为主,轴索损害突出,且为排除性诊断,目前不予考虑;系统性疾病主要是糖尿病,但病史较短,临床表现不符合糖尿病周围神经病特点(感觉障碍突出,疾病晚期出现肌萎缩等);自身免疫性周围神经病(慢性炎性脱髓鞘性多发性神经病)也是排除性诊断,目前不予考虑;该例患者无酒精、化疗药物、重金属等神经毒性物品接触史和长期低氧、寒冷环境暴露史,中毒或恶劣环境导致的周围神经病可能性较小。患者临床表现为典型的“鹤腿”、高弓足,家族史可疑阳性,神经电生理学检查以脱髓鞘改变为主,基因检测显示遗传性周围性运动感觉性神经病(CMT 1型)相关基因PMP22杂合子突变,神经超声支持CMT1型,CMT1型诊断明确。治疗方面,目前对于遗传性周围性运动神经病尚无有效治疗方法,以支持治疗为主。嘱患者康复门诊随诊,在条件允许的情况下可以利用矫形肢具保护肢体、改善活动能力。继续营养神经治疗,避免应用神经毒性药物(如长期使用秋水仙碱、甲硝唑、大剂量维生素B6、长春新碱等)。维持糖尿病饮食,定期监测血糖。
腓骨肌萎缩症(Charcot-Marie-Tooth disease,CMT)
遗传性运动感觉神经病(hereditary motor and sensory neuropathy,HMSN)亦称腓骨肌萎缩症(Charcot-Marie-Tooth disease,CMT),是临床最常见的具有临床表现、神经电生理学和遗传学异质性的慢性进行性遗传性周围神经病变;根据神经电生理学和病理学特征,主要分为脱髓鞘型(CMT1型)和轴索型(CMT2型)。
多数患者于青春期发病(因基因型差异发病时间稍有不同),呈慢性进展性病程,临床主要表现为跨阈步态(step-page gait)、远端肌萎缩,疾病早期以下肢症状显著,呈“鹤腿”或“倒置香槟酒瓶”样表现,继而上肢呈“爪”形手,体格检查可见肢体远端对称性腱反射减弱或消失,本体觉下降或缺失和关节畸形等导致的可疑醉酒步态,多数患者存在轻至中度感觉异常,仅20%~30% CMT1型患者有疼痛主诉(多为骨骼肌症状),部分CMT1A型患者小神经纤维功能障碍突出。该例患者病程中亦出现四肢远端疼痛,可能存在小神经纤维受累,神经功能受损程度可以参考CMT神经病评分。此外,该例患者隐匿起病,慢行进行性病程,对称性周围神经病变(运动和感觉系统均受累),神经电生理学检查以脱髓鞘改变为主,临床拟诊CMT1型。该例患者同时存在感觉障碍、手指和足趾刺痛、糖尿病,在考虑CMT1A型小纤维受累、糖尿病加重CMT临床症状的同时,尚不排除合并糖尿病周围神经病变的可能。然而也有学者认为,CMT合并糖尿病患者发病年龄较晚(35~50岁),发病前多无肥胖表现,临床表现和神经电生理学表现更差,提示可能是CMT的新亚型(CMT先发病或与糖尿病同时发病;临床表现为脱髓鞘型周围神经病,部分出现不典型糖尿病表现;病理改变为脱髓鞘、髓鞘再生和“洋葱皮”样结构;基因型为CMT1型),该例患者临床表现符合该类型,但该类型患者仅见于Yu等报告的包含4个家系在内的30余例患者,且考虑到CMT为单基因遗传性疾病,而多基因相关糖尿病是否与其确切相关目前尚无定论。
神经电生理学检查、神经超声检查和基因检测对明确诊断CMT至关重要。针极肌电图呈现神经源性损害;神经传导速度检查有助于区分髓鞘损伤和轴索损害,传统CMT的临床分型基于临床病理学检查、基因检测和神经电生理学检测(正中神经运动神经传导速度>38m/s为轴索损害,<38m/s为脱髓鞘改变),该例患者正中神经传导速度<38m/s,故归类CMT1型。神经超声是近年新兴的周围神经病变诊断技术,目前逐渐应用于临床。脱髓鞘型CMT(特别是CMT1A型)患者周围神经超声可见神经横截面积(cross sectional area,CSA)增大,提示弥漫性神经增粗,是反复脱髓鞘改变导致施万细胞和神经间质增生的结果。而慢性炎性脱髓鞘性多发性神经病的神经超声显示多灶性块状神经增粗。该例患者神经超声符合典型脱髓鞘改变表现,支持CMT1A型诊断。随着分子生物学的发展,继1991年发现首个CMT致病基因PMP22(17p11.2-12串联重复)以来,相继有60余种CMT致病基因或致病相关基因见诸文献报道。该例患者家族成员调查提示家族史可疑阳性,基因检测显示CMT1A型相关基因PMP22杂合子突变,CMT1型诊断明确;PMP22基因负责编码外周髓鞘型蛋白质(peripheral myelin protein,PMP),该蛋白为周围神经髓鞘重要成分,PMP22基因突变致外周髓鞘型蛋白质拷贝数变异可以造成周围神经髓鞘结构异常,可能是该例患者周围神经病变的发病机制。患者父亲及胞姊可疑四肢远端肌萎缩、肢体偏细,考虑为家族遗传,建议患者直系亲属必要时进行相关基因检测以评估发病风险。基于近年二代测序技术的广泛应用和CMT基因型的异质性特点,基因型与表型之间存在复杂关联的观点也逐渐被认可,传统CMT分类已经不能全面反映出对疾病本质的认识,Mathis等建议采用新的CMT分类标准,根据遗传学表现、临床特点和致病基因进行命名和分类,如AD-CMTde-PMP22dup或AD-AMTax-MFN2,其中包含68种CMT变异型,共计54种已知致病基因和3种未知基因;鉴于其对相关指标和实验室要求较高,目前尚未在临床广泛应用,其可行性尚待进一步检验。
此外,脑脊液指标有助于鉴别特殊类型的周围神经病变,如慢性炎性脱髓鞘性多发性神经病出现经典的“蛋白-细胞分离”现象。值得注意的是,某些CMT类型(如CMT1型)脑脊液蛋白定量可轻度升高,该例患者脑脊液蛋白定量升高,故应结合其他临床和辅助检查综合评价。尽管周围神经组织活检术提供的信息有限且常造成不可逆性损害,临床应用受到限制,但近年有学者提出,通过皮肤组织活检术了解皮肤髓鞘的病理改变,有助于获得更多信息(如Meissner小体数量、有髓纤维末梢密度和直径、Ranvier结间和结内长度),从而明确诊断。
尽管针对CMT致病机制开展的药物研究在动物实验中取得成果,但目前尚无药物成功通过临床试验并证实对疾病有缓解作用,因此,CMT的治疗仍以支持治疗为主。多种矫正治疗可以改善患者活动受限和提高生活质量,如脚踝矫正术、拇对掌夹板等,严重脊柱侧凸患者可以采取手术治疗。适当的康复锻炼不会加重CMT患者的运动障碍,是一种安全、有效的治疗手段。该例患者明确诊断后在康复科门诊随诊,进行肢体康复训练,康复情况良好。
综上所述,CMT迄今尚无特效治疗方法,因此对于临床疑诊CMT的患者,详细的病史询问,包括家族史、中毒史和药物接触史,血液生化检查,包括免疫指标、M蛋白相关标志物、甲状腺功能试验、血糖、肿瘤学标志物等均有助于排除获得性周围神经病变;神经电生理检查、神经超声有助于临床分类;基因检测有助于进一步明确疾病类型。
[1]Patzko A,Shy ME.Charcot-Marie-Tooth disease and related genetic neuropathies.Continuum (Minneap-Minn),2012,18 (1):39-59.
[2]Tazir M,Hamadouche T,Nouioua S,et al.Hereditary motor and sensory neuropathies or Charcot-Marie-Tooth diseases:an update.J Neurol Sci,2014,347 (1-2):14-22.
[3]Yiu EM,Brockley CR,Lee KJ,et al.Peripheral nerve ultrasound in pediatric Charcot-Marie-Tooth disease type 1A.Neurology,2015,84 (6):569-574.
[4]Hoyle JC,Isfort MC,Roggenbuck J,et al.The genetics of Charcot-Marie-Tooth disease:current trends and future implications for diagnosis and management.Appl Clin Genet,2015,8:235-243.
[5]Manganelli F,Nolano M,Pisciotta C,et al.Charcot-Marie-Tooth disease:New insights from skin biopsy.Neurology,2015,85 (14):1202-1208.
[6]Corrado B,Ciardi G,Bargigli C.Rehabilitation Management of the Charcot-Marie-Tooth Syndrome:A Systematic Review of the Literature.Medicine (Baltimore),2016,95 (17):e3278.
[7]徐银燕,张江涛,牛婧雯,等.进行性四肢麻木无力4年.中国现代神经疾病杂志,2017,017,17(1):74-77.









