


 收藏
收藏 已收藏
已收藏(一)病例信息
【病史】
女性患者,53岁,家庭妇女,因间断咳嗽、活动后呼吸困难10年,呼吸困难进行性加重5年,于2013年5月入院。患者自2003年1月起无明显诱因出现咳嗽,以干咳为主,偶尔咳少量白色黏痰,活动后呼吸困难,无胸痛、憋喘、咯血、盗汗等;2003年7月开始咳嗽、呼吸困难加重,出现咯血1次(约5ml),于当地医院就诊。胸部CT显示纵隔及肺门多发淋巴结肿大,双肺弥漫性分布磨玻璃样小叶中心型结节伴多发小薄壁空腔(肺气囊)。全麻下行纵隔镜淋巴结活检,术中见颈部及气管右前(主动脉弓上缘平面)多枚淋巴结肿大。淋巴结病理报告提示淋巴结反应性增生。当地医院诊断为结节病,给予口服泼尼松龙(30mg/d)联合环磷酰胺(200mg/d,1个月)治疗。患者用药后,症状减轻,遂逐渐减少泼尼松龙剂量(每2周减10mg),减至10mg/d时症状又加重,以夜间为著,伴有胸闷,不能平卧入眠。2004年7月,患者再次住院治疗。口服泼尼松50mg/d 2个月后,逐渐减量至15~20mg/d维持,患者症状减轻且较稳定,能耐受一般日常活动。2008年10月,患者症状再次加重,活动耐力明显下降,联合甲氨蝶呤10mg/w治疗2个月,咳嗽、咳痰症状略有减轻,但活动后呼吸困难无缓解。近1年,患者呼吸困难呈进行性加重,活动明显受限,稍动即出现呼吸困难,采取长期家庭氧疗,且多次住院治疗。发病以来,除了被诊断为结节病外,还曾疑诊肉芽肿性血管炎和肺淋巴管平滑肌瘤病等。
患者既往有子宫平滑肌瘤剔除术史、右髋部外伤史,否认糖尿病、高血压病、冠心病等病史,否认家族遗传性疾病病史。
【体格检查】
体温36.8℃,血压122/82mmHg,心率96次/分,呼吸28次/分;口唇发绀,全身浅表淋巴结未触及肿大;双肺呼吸音低,未闻干湿啰音;心界不大,各瓣膜听诊区未闻病理性杂音;肝-颈静脉回流征阴性,腹部及神经系统查体无异常发现;双下肢无水肿,无杵状指。
【实验室检查】
血气分析(室内空气):pH 7.43,PaO2 62mmHg,PaCO2 33mmHg,HCO3- 21.9mmol/L,SaO2% 92%。
血常规:WBC 9.75×109/L,N% 73%,L% 17%,Mo% 10%,RBC 4.31×1012/L,Hb 105g/L。
血沉:102mm/1h。
生化:ALB 36.5g/L,球蛋白(globulin,GLB)65.7g/L,钙磷等电解质水平正常。血清血管紧张素转换酶(SACE)26U/L。
肺癌相关肿瘤标志物均阴性,HBV、HCV和HIV均阴性。
凝血功能正常,血清D-二聚体正常。
免疫相关检查:血清自身抗体(抗Sm、抗SSA、抗SSB、抗Jo-1抗体、抗Sc1-70抗体等)均阴性。免疫球蛋白:IgG 45.9g/L,IgA 8.5g/L,IgM 3.42g/L,补体C3、C4正常。
病原学检查:痰细菌培养、真菌培养、痰涂片查抗酸杆菌均阴性,PPD试验阴性。
甲状腺功能正常;脑钠肽(BNP)正常。
【影像学检查】
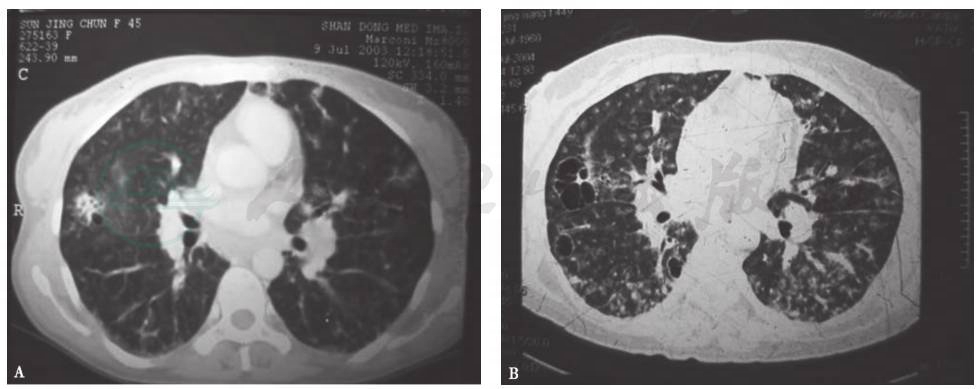
2003年7月胸部CT显示纵隔及肺门多发淋巴结肿大,双肺弥漫性分布磨玻璃样小叶中心型结节,边界不清,可见散在多发小薄壁空腔(肺气囊)(图1)。9年后(2012年)复查胸部CT显示纵隔及肺门多发肿大淋巴结,较前(2003年)无明显变化,但多发肺大疱更为明显(图2)。
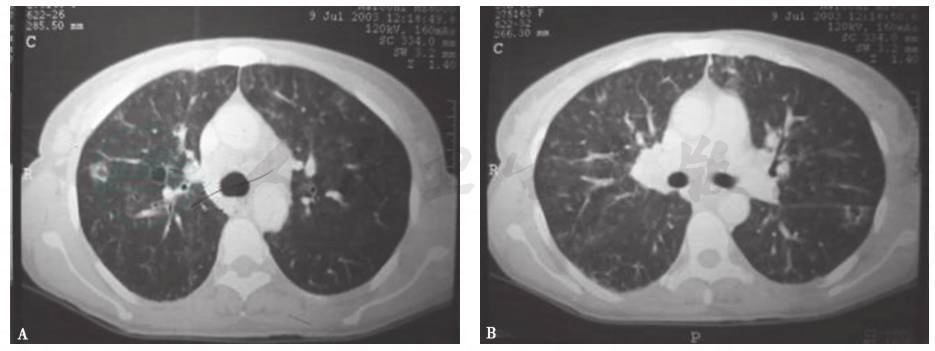
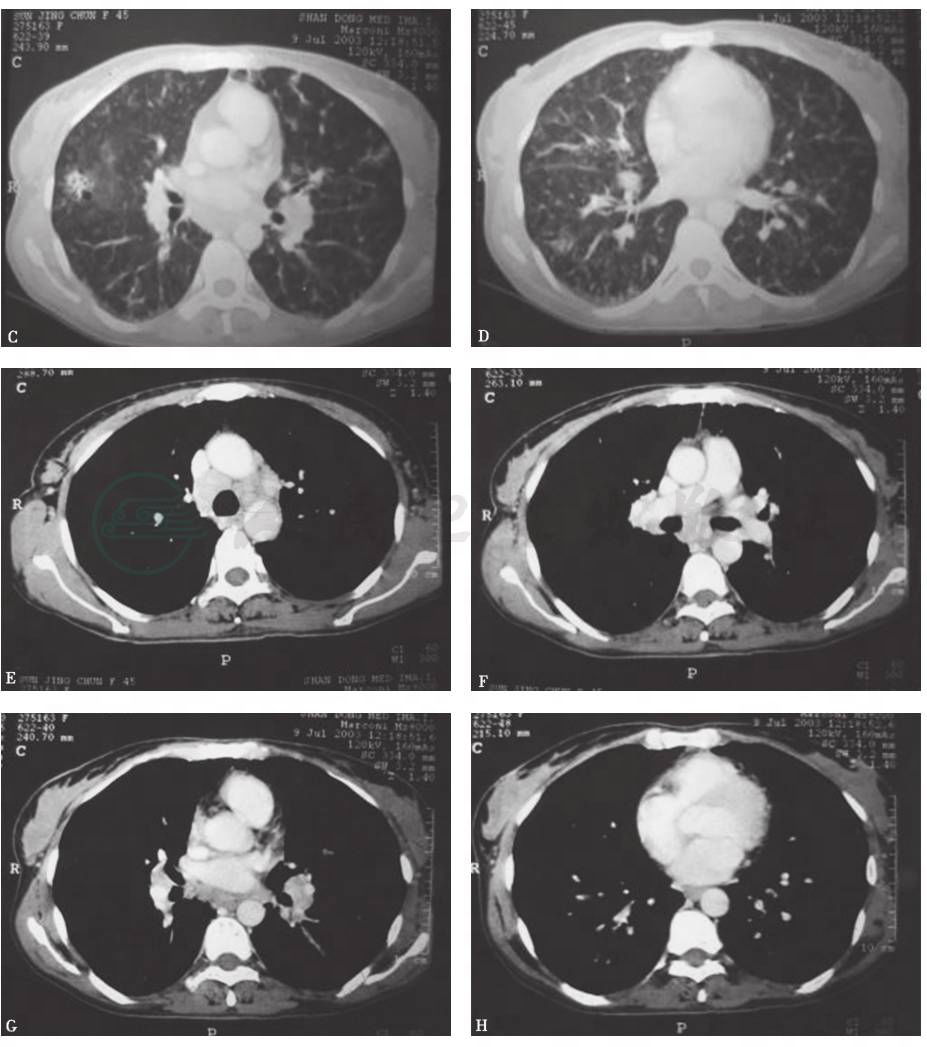
图1 2003年胸部CT表现
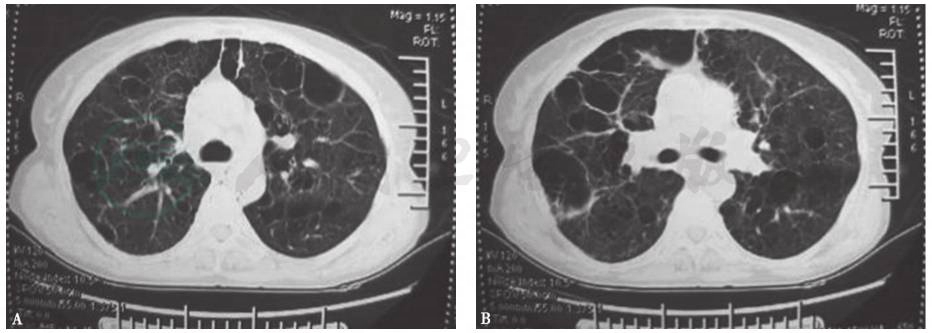
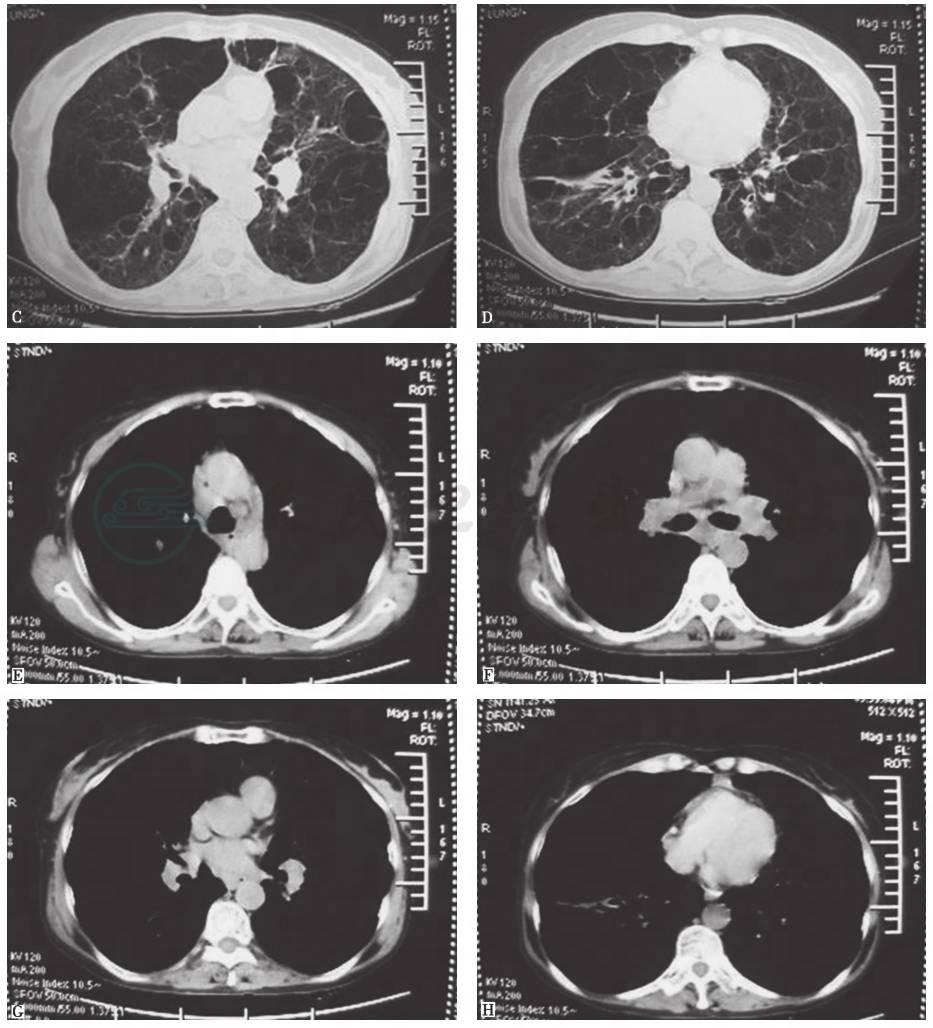
图2 2012年胸部CT表现
【肺功能检查】
TLC 0.54L,FEV1/FVC 0.44,FEV1/pre 0.29,弥散/通气比 0.44。
【心脏彩超】
心脏结构及室壁运动未见明显异常。
(二)临床思辨
【临床特点】
1.患者为中老年女性,病程呈慢性,病情进行性加重。
2.主要症状和体征为反复咳嗽,进行性加重的劳力性呼吸困难,口唇发绀,双肺呼吸音低,未闻干湿啰音。
3.实验室检查显示多克隆免疫球蛋白升高、ESR升高;自身抗体谱、肿瘤标志物、PPD试验、HIV等阴性。
4.肺功能检查提示重度阻塞性通气功能障碍合并重度弥散功能障碍。
5.胸部CT早期表现为双肺弥漫性分布的磨玻璃样小叶中心型结节,边界不清,并可见散在小肺气囊,合并纵隔及肺门多发淋巴结肿大,其中纵隔及肺门肿大淋巴结与2003年相比无明显变化。
6.糖皮质激素、免疫抑制剂等治疗反应欠佳。
【思辨要点】
本病例以干咳、进行性加重的劳力性呼吸困难为主要临床表现,纵隔淋巴结活检诊断为结节病,但在使用糖皮质激素情况下病情仍持续进展。
1.本例患者出现呼吸困难进行性加重的原因是什么?
呼吸困难在临床极为常见,其病因错综复杂,主要可分为肺源性呼吸困难(如阻塞性、限制性通气障碍和肺血管疾病)及肺外呼吸困难(如心源性、神经肌肉病变等)。本例患者出现渐进性劳力性呼吸困难,肺功能检查提示重度阻塞性通气功能障碍伴弥散功能减低,胸部CT见双肺多发肺大疱,未见肺动脉段增宽、胸腔积液等改变,超声心动图等未提示心脏结构改变或射血分数减低、肺功能高压,BNP正常。分析其呼吸困难加重的原因,可能主要与病变进展,肺实质破坏、肺有效通气面积明显减少相关。
2.对于本例患者,结节病诊断的依据是否充分?
糖皮质激素一直是结节病治疗的首选药物,对缓解症状、减少肉芽形成有一定疗效。多数患者对糖皮质激素反应良好,但也有少数患者反应不佳或不能耐受激素不良反应,对于后者可考虑使用免疫抑制剂。
本例患者在持续应用激素情况下,临床症状仍缓慢进展,病程中曾短时间应用环磷酰胺等药物,但症状仍无改善,因此结节病诊断依据不足,需通过病理检查进一步排除。
(一)临床信息
【实验室检查】
详细回顾患者发病11年来的诊疗资料,异常的实验室检查项目主要为持续的多克隆高免疫球蛋白血症以及血沉增快;肿瘤标志物、PPD试验、自身抗体等指标则持续为阴性(表1)。
表1 发病11年间血清总蛋白、白蛋白、球蛋白及其亚型和血沉一览表
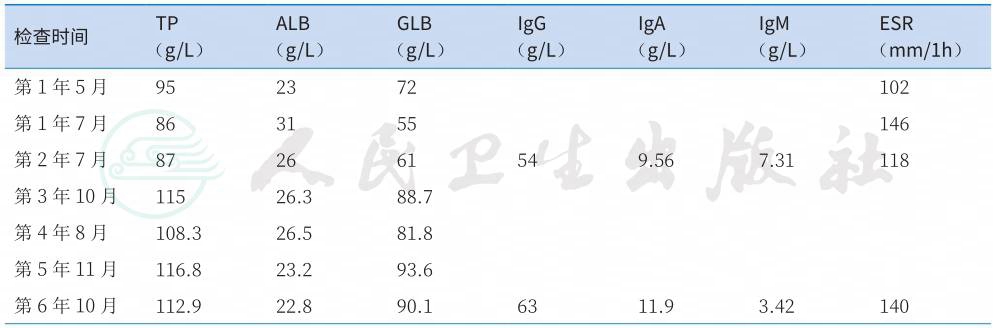
续表
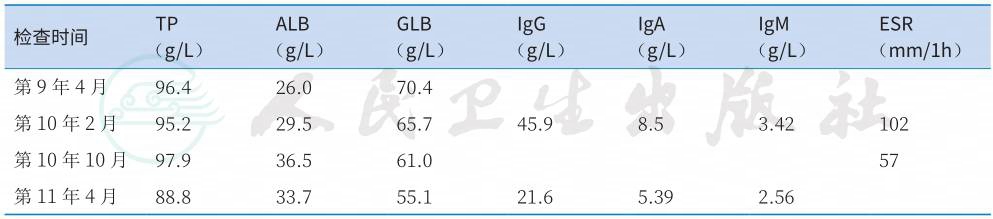
最初就诊时间:2003年1月
【肺功能检查】
患者肺功能检查显示混合性通气功能障碍[初期以限制性通气功能障碍为主,后期则以阻塞性通气功能障碍(重度)为主],弥散功能呈进行性减退(表2)。
表2 发病11年期间肺功能演变
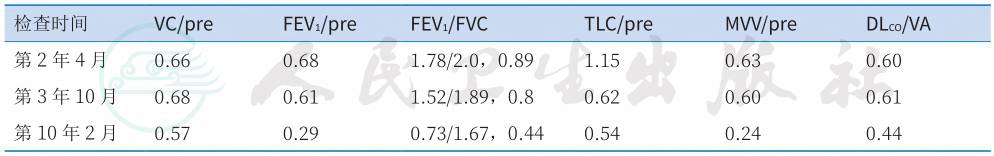
检查时间 VC/pre FEV1/pre FEV1/FVC TLC/pre MVV/pre DLCO/VA |
第2年4月 0.66 0.68 1.78/2.0,0.89 1.15 0.63 0.60 第3年10月 0.68 0.61 1.52/1.89,0.8 0.62 0.60 0.61 第10年2月 0.57 0.29 0.73/1.67,0.44 0.54 0.24 0.44 |
引自:主编:.呼吸系统疑难病例诊疗辨析.第1版.ISBN:978-7-117-26415-0
【影像学检查】
患者患病11年间胸部CT(图3、图4)始终可见纵隔及肺门肿大淋巴结,肺实质内开始为双肺弥漫性分布的磨玻璃样小叶中心型结节,边界不清,并可见散在薄壁囊腔(肺气囊),之后薄壁囊腔逐年增多,小结节和磨玻璃影逐渐减少,未见牵拉性支气管扩张、蜂窝等纤维化改变。
A.第1年;B.第2年;
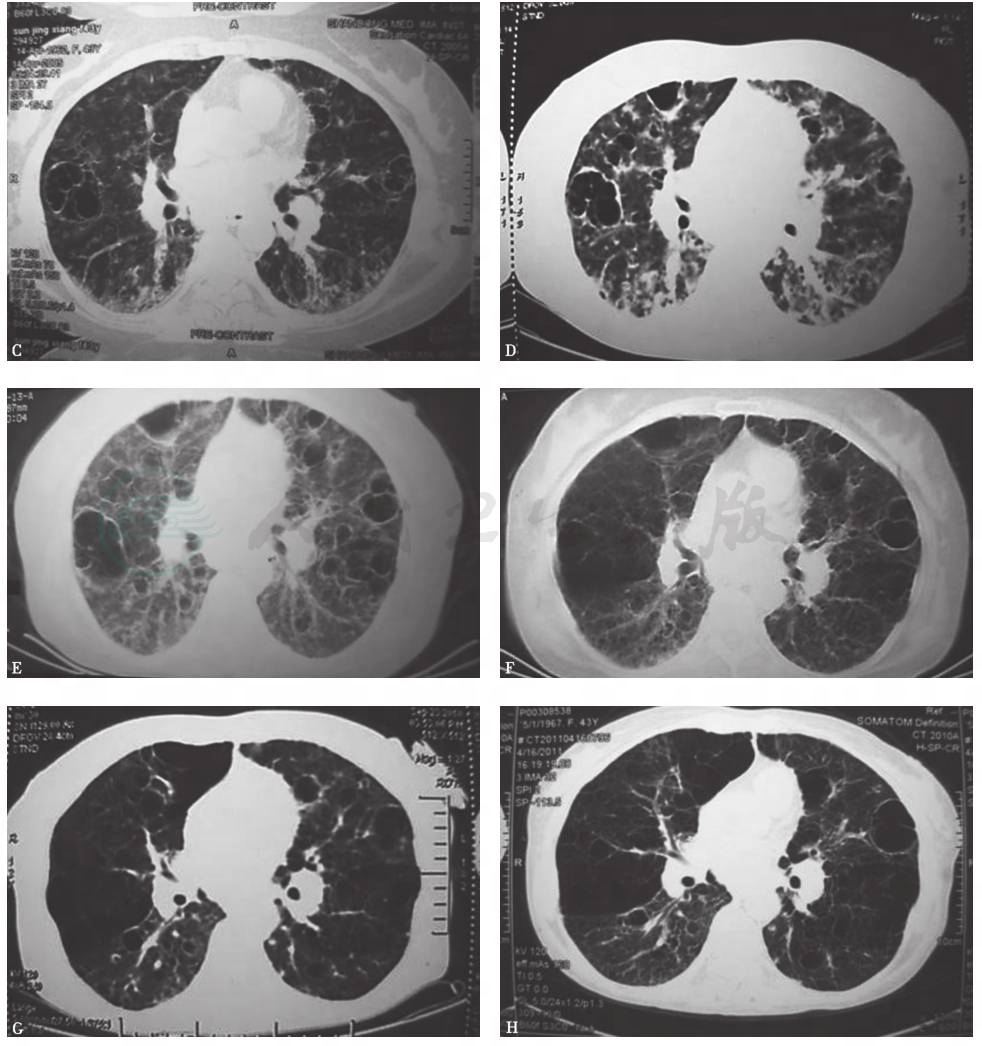
C.第3年;D.第4年;E.第6年;F.第7年;G.第8年;H.第9年;
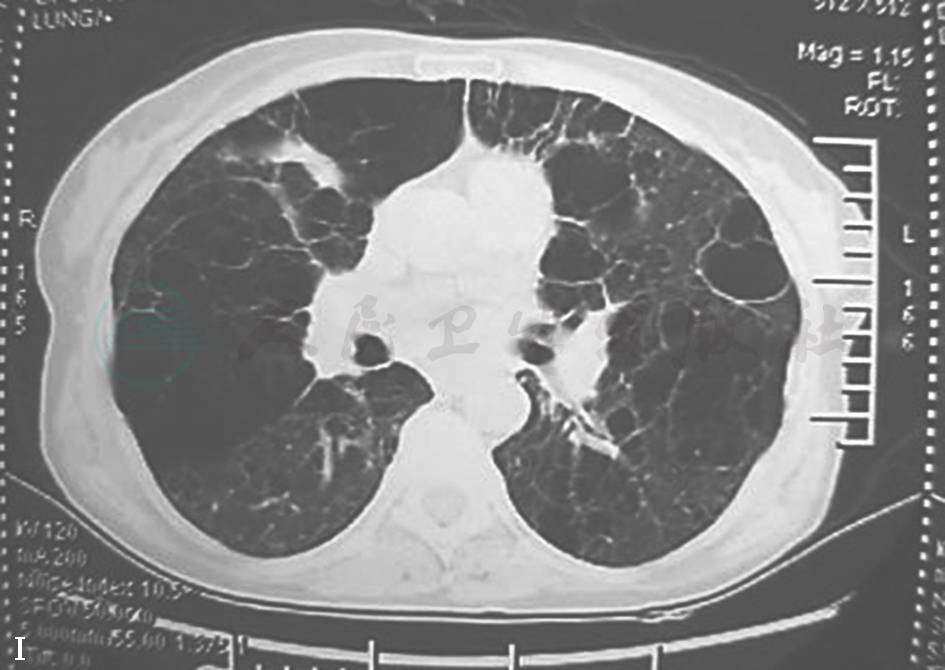
I.第10年
图3 2003—2012年胸部CT肺窗演变过程
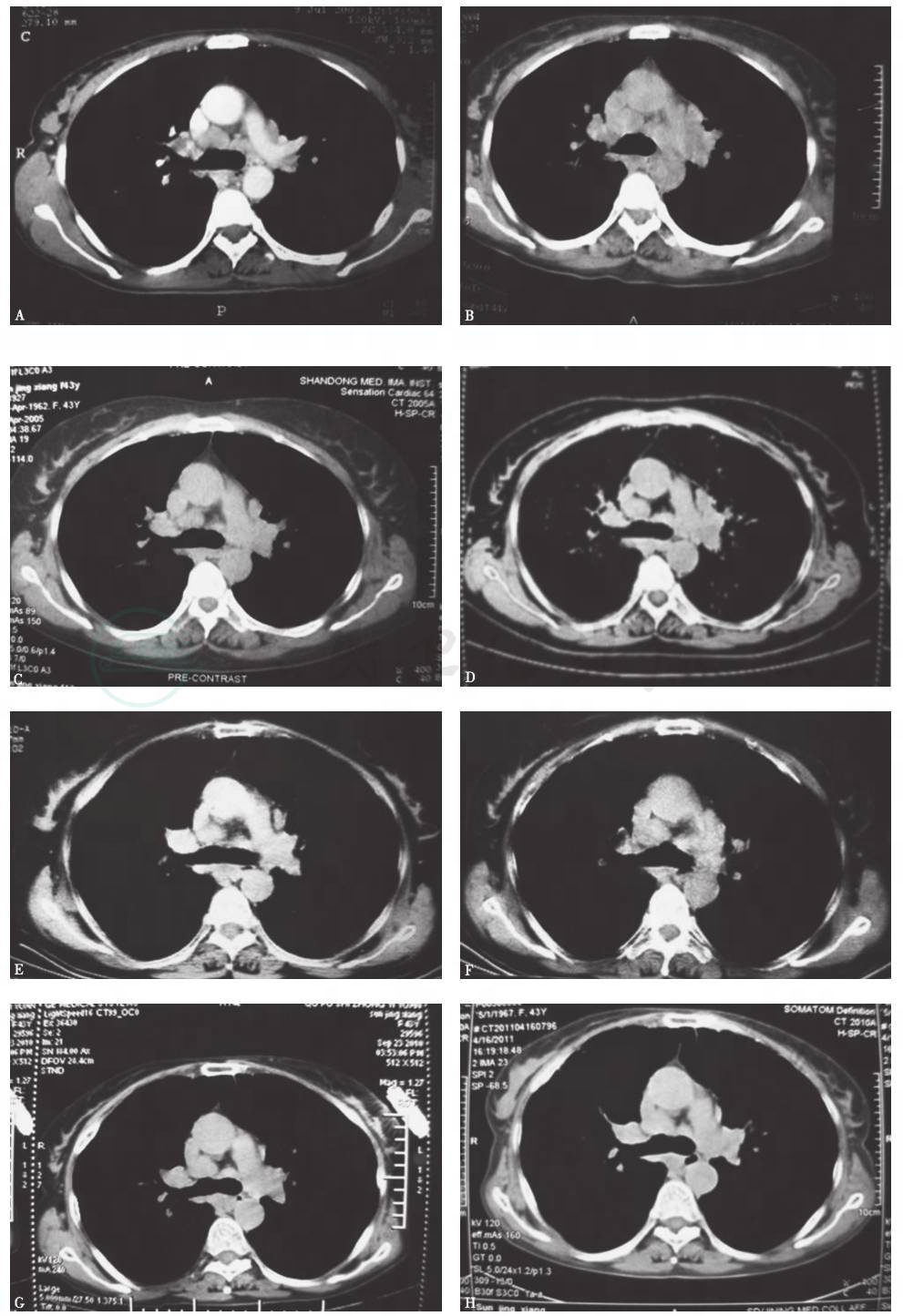
A.第1年;B.第2年;C.第3年;D.第4年;E.第6年;F.第7年;G.第8年;H.第9年;
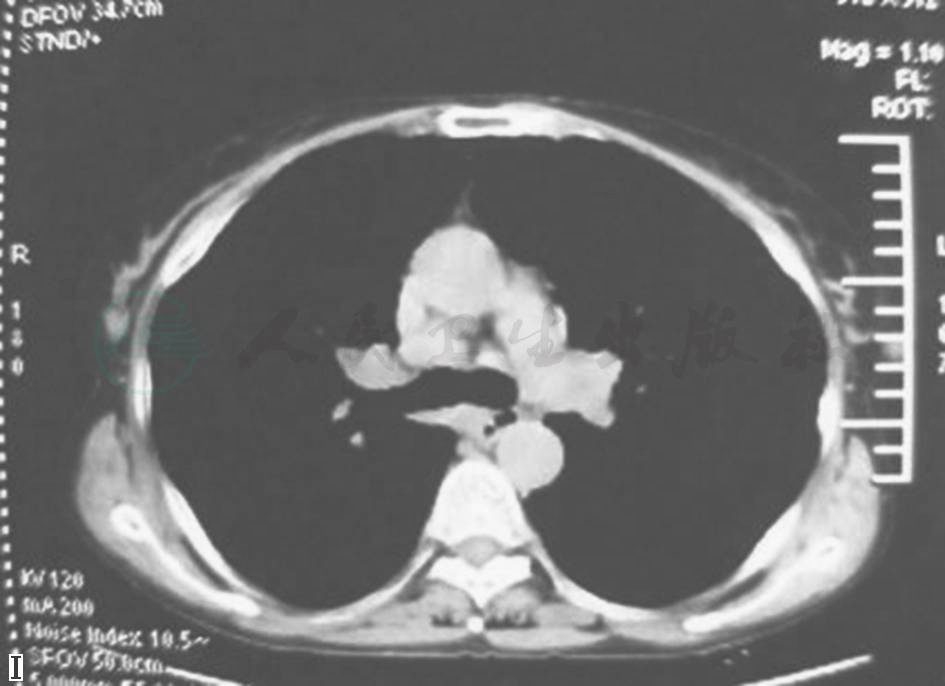
I.第10年
图4 2003—2012年胸部CT(纵隔窗)演变过程
(二)临床思辨
根据现有临床资料,对于本病例,需要做哪些鉴别诊断?
上述临床资料显示,本病例的基本特点为:慢性起病、病程长,病变局限在肺内,血沉快,多克隆免疫球蛋白水平增高;影像学表现为肺内弥漫性小结节和磨玻璃影、薄壁囊腔,随时间推移发展为双肺多发肺大疱,肺实质破坏明显,导致渐进性呼吸困难,最终发展至呼吸衰竭。
针对上述临床特点,须对下列疾病进行鉴别诊断:
(1)淋巴管平滑肌瘤病(LAM)
育龄期妇女若双肺出现多发肺气囊,需要考虑该病。LAM以不典型平滑肌细胞(LAM细胞)过度增生为特征,可因小气道平滑肌细胞异常增生堵塞远端气道及淋巴管而出现气胸、乳糜胸等改变。但LAM的影像学改变并不包括肺内磨玻璃样渗出性小结节,并且不伴有免疫球蛋白异常增加,与本病例情况不符。
(2)肺朗格汉斯细胞组织细胞增多症(PLCH)
也可出现双肺多发薄壁囊腔。该病90%发生于吸烟者,其囊状影分布不均且大小不等,常伴有多发性结节,个别患者可有肺门和纵隔淋巴结肿大。但PLCH的结节状和囊性病变主要分布在肺上部区域,双下肺肋膈角病灶少见,并且不伴有免疫球蛋白的异常增加,与本病例情况不符。
(3)IgG4相关性肺疾病(IgG4-related disease,IgG4-RD)
可有多克隆性高免疫球蛋白血症,其影像学可表现为实性结节影、磨玻璃样改变或蜂窝肺。Inoue等根据CT特点把IgG4相关性肺疾病分为实性结节型、多发圆形磨玻璃影型、肺泡间质型和支气管血管束型4种主要类型。其中,支气管血管束型和肺泡间质型的CT表现与肺淋巴组织增生性疾病等难以鉴别,本例患者血清IgG4正常,不符合该病特征,但还需要对淋巴结组织进行IgG和IgG4组化染色,以进一步排除。
(4)淋巴细胞间质性肺炎(LIP)
HRCT可显示边界不清的小叶中央磨玻璃影样结节和胸膜下小结节、支气管血管束增厚等,病变多分布于下叶,80%患者可出现薄壁囊腔;病理特征为弥漫性肺间质致密淋巴细胞浸润。本病例的影像学与LIP相似。但是,特发性LIP很少同时合并有纵隔淋巴结肿大和免疫球蛋白异常增加。
(5)多中心型Castleman病(CD)
属于淋巴组织增生性疾病的一种,可以出现纵隔及肺门淋巴结肿大;由于浆细胞在肿大淋巴结异常表达,可导致免疫球蛋白异常增高,血沉也相应增快;病变累及肺时,其病理组织学表现为LIP。本例患者的临床表现、影像学表现和实验室检查指标的异常改变均符合CD的特点,但确诊还需要依赖病理及免疫组织化学检测。
(6)淋巴瘤
也可出现多发淋巴结肿大,常伴持续或周期性发热、消瘦、脾大等症状。本病例病程迁延时间长,无慢性发热,未出现肺外淋巴结受累,故诊断该病依据不足。
(7)其他
纵隔和肺门多发淋巴结肿大还可能发生于肺癌和肺结核。本病例病程迁延时间长,未出现结核中毒症状;胸部CT检查显示结节影和磨玻璃影逐渐减少,未出现占位性病变;肿瘤标志物正常;痰涂片未查到抗酸杆菌,PPD试验阴性。上述表现不支持肺癌和肺结核诊断。对于其他全身性疾病,如结缔组织疾病等,目前没有相应诊断依据。
(一)临床信息
【实验室检查】
免疫球蛋白 亚类:IgG1 13.33g/L,IgG2 4.14g/L,IgG3 0.41g/L,IgG4 0.11g/L。
球蛋白κ轻链16g/L,λ轻链11.4g/L。
【组织病理学】
重新对本例患者纵隔淋巴结的病理切片进行组化染色可见:HE染色显示淋巴结正常结构大致存在,淋巴组织增生活跃,可见滤泡结构,滤泡间有大量成熟浆细胞浸润;免疫组化显示CD20、CD3、CD38、CD138和IgG部分细胞呈阳性,Ki-67增殖指数5%阳性,仅少数细胞呈IgG4阳性(IgG4阳性浆细胞数<10个/HP,IgG4/IgG<10%)(图5)。
结合病史和血清IgG4水平,本病例可排除IgG4-RD,诊断为多中心性浆细胞型Castleman病。
最后诊断:多中心性浆细胞型Castleman病。
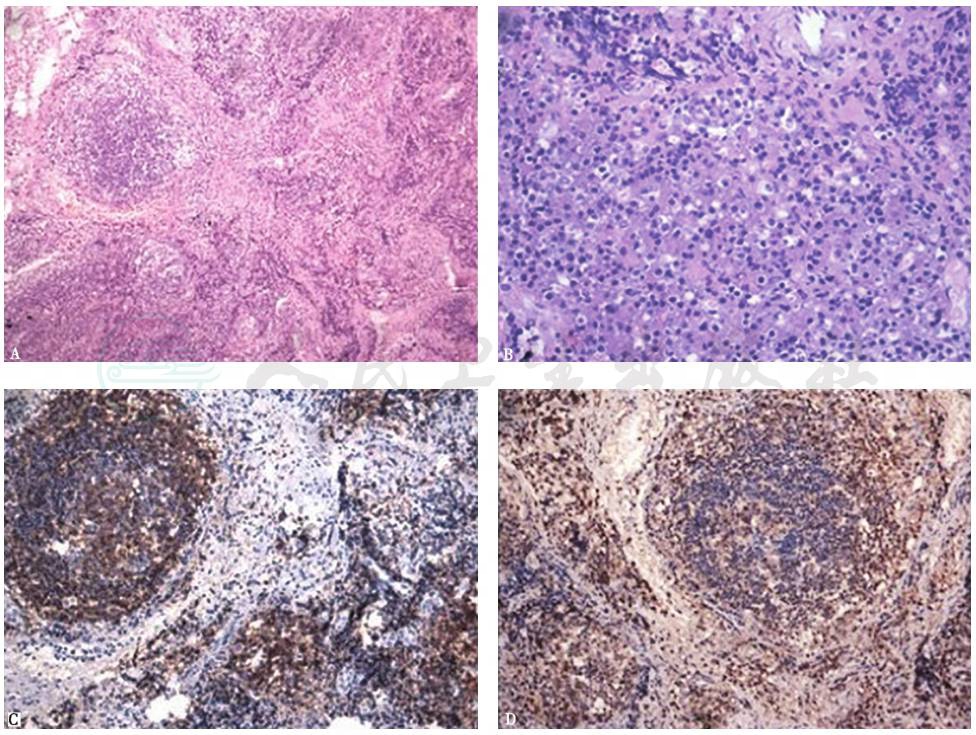
HE染色可见淋巴结结构、滤泡结构,其间有大量分化成熟的浆细胞(A.HE染色,100×;B.HE染色,400×);组化染色CD20(C.200×)、CD3(D.200×)
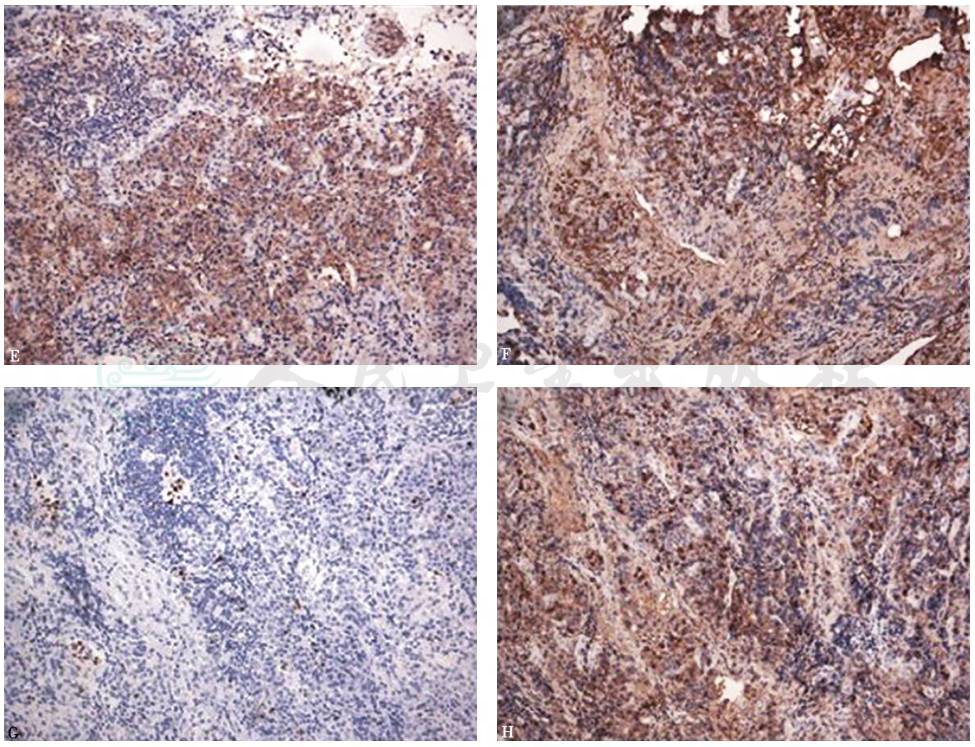
CD138(E.200×)和IgG部分细胞呈阳性(F.200×),Ki-67增值指数5% (G.200×),仅少数细胞呈IgG4阳性(H.200×)
纵隔淋巴结病理表现 图5
(二)临床思辨
Castleman病(CD)是一组少见的非典型淋巴组织增殖性疾病,又称为巨大淋巴结增生症、血管淋巴滤泡增生症、血管瘤样淋巴结增生症和淋巴样错构瘤等,由Castleman等在1956年首次报道。该病突出的临床表现为无痛性淋巴结肿大,大多数病例表现为局限性淋巴结病变,最常侵犯纵隔淋巴结,颈部、后腹壁、腋窝及盆腔等部位的淋巴结也可被侵犯,含有淋巴组织的器官(如肺、喉、腮腺、胰腺、肌肉等)可被累及。其肿大淋巴结的组织病理学表现为:①肿大淋巴结结构基本保持完整;②滤泡增生明显;③血管增生。CD可进一步分为透明血管(hyaline vascular,HV)型、浆细胞(plasma cell,PC)型和混合型。HV型突出表现为滤泡血管呈玻璃样变,伴滤泡生发中心萎缩;PC型则突出表现为滤泡间质中浆细胞浸润增多,伴滤泡生发中心增生;混合型具有二者的共同表现。
临床上多将Castleman病按累及范围分为局限型Castleman病(unicentric Castleman's disease,UCD)和多中心型Castleman病(multicentric Castleman's disease,MCD)。UCD表现为孤立淋巴结或纵隔内某一组淋巴结肿大,临床多无症状,90%以上为透明血管型,多可手术切除,预后较好。MCD表现为一组以上的淋巴结或淋巴结外器官受累,通常有低热、盗汗、乏力等全身症状,实验室检查提示贫血、血沉加快、多克隆性高免疫球蛋白血症等,绝大多数为浆细胞型,手术切除困难,预后较差。目前,多数研究认为,IL-6在CD发生发展过程中起重要作用,而HIV或HHV-8等病毒感染与MCD预后关系密切。
对于局限于肺内的MCD,其肺实质病理组织学表现为B淋巴细胞和大量浆细胞在肺实质内的浸润;HRCT可见肺门和(或)纵隔巨大淋巴结增生,肺实质改变类似LIP,表现为边界不清的小结节、磨玻璃影、肺气囊、小叶间隔增厚以及外周支气管血管束增粗、双侧少-中量胸腔积液等。Johkoh等报道一组(12例)胸内MCD,影像学表现为肺实质广泛浸润,全肺均受累,弥漫分布的小叶中心性模糊小结节、磨玻璃样改变、散在肺气囊等改变。薄壁气囊可能是由支气管或细支气管周围淋巴细胞间质性肺炎引起气道不全性阻塞而形成。
本例CD患者病程长达十余年,影像学资料非常完整,有助于我们对本病肺部病变演变过程深入认识。其胸部CT表现始终可见纵隔及肺门淋巴结肿大,肺实质内早期为双肺弥漫性分布的小结节和磨玻璃高密度病灶,边缘较模糊,可见多发散在的肺气囊;病程进入第5年后,肺实质内肺气囊进行性增多,而双肺弥漫性分布的小结节和磨玻璃影病灶明显减少;发病6年(2009年)后,肺内的基本改变为多发肺气囊和肺大疱。患者初期以限制性通气功能障碍为主,后期则以重度阻塞性通气功能障碍为主,而弥散功能呈进行性减退,其肺功能变化与肺内肺气囊增加的影像学改变相符。
MCD没有标准治疗方案,既往多采用激素和(或)化疗,常采用CHOP方案(环磷酰胺+多柔比星+长春新碱+泼尼松),但疗效存在争议。
有研究显示,是否合并病毒感染显著影响患者预后,因此诊断明确为MCD后,应进行HIV及HHV-8病毒检测。对于合并HIV/HHV-8感染者,可考虑使用抗IL-6单克隆抗体或IL-6受体抑制剂,无法获取IL-6单抗或受体抑制剂时可考虑使用抗CD20单克隆抗体(利妥昔单抗)。对于合并HIV/HHV-8感染者,则应同时启动抗病毒治疗(如更昔洛韦)。其他治疗选择还包括硼替佐米、沙利度胺等,也显示一定疗效。
本例患者基础情况较差,病程迁延时间长达11年,可能部分得益于病初阶段给予糖皮质激素联合免疫抑制剂治疗。
Castleman病是一组少见的非典型淋巴组织增殖性疾病,临床诊断困难。对于多中心型并发弥漫性间质肺病患者,如果发现同时伴有高免疫球蛋白血症,高度疑诊MCD,需要临床医师仔细甄别临床每一个异常环节和指标。正确诊断的确立往往是一个循环往复的认识过程,需要对之不断认识-否定-再认识-再否定,对少见的“孤儿性”肺病而言,更是如此。
(郑雅莉 张洋 高占成)
1.Inoue D,ZenY,AboH,et al.Immunoglobulin G4-related lung disease:CT findings with pathologic correlations.Radiology,2009,251:260-270
2.Iyonaga K,Ichikado K,Muranaka H,et al.Multicentric Castleman's disease manifesting in the lung:clinical,radiographic,and pathologic findings and successful treatment with corticosteroid and cyclophosphamide.Intern Med,2003,42:182-186.
3.Castleman B,Iverson L,Menedez VP.Localized mediastinal lymphnode hyperplasis resembling thymoma.Cancer,1956,9:822-830.
4.Talat N,Schulte KM.Castleman's disease:systematic analysis of 416 patients from the literature.The oncologist,2011,16(9):1316-1324.
5.Johkoh T,Muller N,Ichikado K,et al.Intrathroacic multicentric castleman disesae:CT findings in 12 patients.Radiology,1998,209:477-481.
6.Desar SR,Nicholson AG,Stewart S,et al.Benign pulmonary lymphocytic infiltration and amyloidosis :computed tomographic and pathologic features in three cases.J Thorac Imaging,1997,12:215-220.









