


 收藏
收藏 已收藏
已收藏(一)病例信息
【病史】
女性患者,59岁,因咳嗽、咳痰伴活动后胸闷、气喘2个月入院。患者自诉于2个月余前无明显诱因出现咳嗽、咳痰(白黏痰,量不多),伴有活动后胸闷、气喘,双肩关节疼痛,上举困难,下蹲后站起乏力,间断发作,于当地医院就诊,查胸部CT示两肺间质性改变,经哌拉西林舒巴坦抗感染及化痰等治疗,咳嗽、咳痰较前好转,但活动后气喘仍明显。患者来我院进一步诊治,门诊以“间质性肺炎”收住入院。发病以来,患者有口干,无眼干,无皮疹、口腔溃疡、光过敏,无发热、畏寒,无胸痛、咯血,精神、食欲、睡眠一般,大小便正常,体重无明显变化。
患者既往身体健康,无烟酒嗜好,务农,家族史无特殊。
【体格检查】
体温36.3℃,心率79次/分,呼吸22次/分,血压113/78mmHg;神志清,精神可,口唇无发绀;颈软,颈静脉无怒张;气管居中,两肺呼吸音粗,两下肺可闻Velcro音;心律齐,各瓣膜区未闻病理性杂音;腹平软,无压痛反跳痛,肝、脾肋下未触及;肝肾区无叩痛,移动性浊音阴性;双肘关节皮肤干燥、脱屑;双下肢无水肿,生理反射存在,病理反射未引出。
【影像学检查】
胸部CT(外院):两肺间质性改变。
(二)临床思辨
【临床特点】
1.患者为中年女性,咳嗽、咳痰伴活动后胸闷、气喘2个月,有肢体活动受限。
2.两肺呼吸音粗,两下肺可闻Velcro啰音,心律齐,双肘关节皮肤干燥、脱屑,双下肢无水肿。
3.胸部CT示两肺间质性改变。
4.抗细菌治疗效果不佳。
【思辨要点】
1.对于本病例,如何判断病因类别?
本例患者是中年女性,临床表现为咳嗽、咳痰及气喘,无特异性,肺炎、慢性阻塞性肺疾病、咳嗽变异性哮喘、支气管扩张等均可引起。感染性疾病是致病微生物(如细菌、真菌、病毒等常见病原体及其毒素或肺炎支原体、衣原体和结核分枝杆菌等非特异性病原体)感染人体后,病原体侵入部位可首先出现症状,如果病原体侵入呼吸系统,临床表现可有咳嗽、咳痰、发热及气喘等。发热(尤其是高热)对于感染性疾病的诊断有重要意义。通常情况下,免疫功能低下患者感染时可以无发热,如长期应用免疫抑制剂、患糖尿病和老龄等。本例患者是免疫功能状态正常的中年女性,在2个月病程中始终无发热,故由感染性病因所致可能性很小。而且患者在入院前2个月期间病情相对稳定,呈亚急性和慢性病程,更不支持活动性感染。患者胸部HRCT的特点是双肺散在磨玻璃影、网状影、牵拉性支气管扩张、胸膜下线,病变部位以胸膜下及双下肺为主。这样的影像学特点多见于特发性肺纤维化(IPF)、结缔组织疾病相关间质性肺疾病(connective tissue disease associated interstitial lung disease,CILD)、隐源性机化性肺炎(COP)、脱屑性间质性肺炎(DIP)、过敏性肺炎(HP)、放射性肺炎(RP)等非感染性肺部疾病,也可见于肺孢子菌肺炎及病毒性肺炎。肺孢子菌肺炎主要见于艾滋病患者及免疫功能受损患者,不符合本例患者的状态和临床表现。而病毒性肺炎的特点是急性起病、进展迅速,或导致急性呼吸窘迫综合征(ARDS)或趋于自限,病程不会迁延,也与本例患者的临床过程不相符。
综上所述,本患者咳嗽、咳痰、活动后胸闷气短,HRCT呈磨玻璃影,考虑CILD可能,进一步确诊需要结合临床、影像、病理(必要时)检查结果。
2.如何对弥漫性肺疾病不同病因进行鉴别诊断?
根据以上分析,基本明确本例患者肺部病变为非感染性的弥漫性肺疾病,接下来应结合患者HRCT特点,在可能的CILD病因中进行鉴别诊断,帮助选择进一步检查的方法,以便确诊。
(1)特发性肺纤维化(IPF)
是一种原因不明的慢性致纤维化性间质性肺炎,组织病理学表现为普通型间质性肺炎(UIP),病变局限于肺部。一般认为,环境刺激、病原体感染及自身免疫异常等均可成为本病的致病因子。IPF一般慢性起病,临床表现为干咳、呼吸困难等。其典型的HRCT改变表现为网格影,伴有斑片状改变,以外周和胸膜下分布为主,蜂窝样改变明显,并见牵拉性支气管扩张征象,磨玻璃影相对比较少见(图1)。肺功能表现为限制性通气功能障碍。典型IPF依据临床表现及典型HRCT改变即可诊断,不典型IPF需要肺活检病理明确诊断。目前IPF的治疗主要以吡非尼酮或尼达尼布为主,糖皮质激素或激素联合免疫抑制剂疗效不佳。
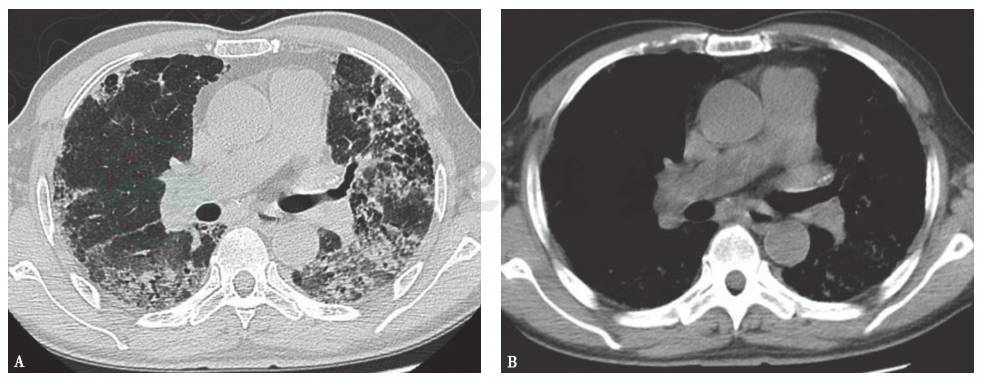
图1 特发性肺纤维化胸部CT表现
男性患者,73岁,咳嗽伴气喘2年,加重1个月。胸部CT可见双肺弥漫间质性改变,小叶间隔和小叶内间隔增厚,病变主要分布于胸膜下,并见牵拉性支气管扩张伴蜂窝样改变。临床诊断为特发性肺纤维化
(2)隐源性机化性肺炎(COP)
指没有明确致病原(感染)和其他临床伴随疾病(如结缔组织疾病等)情况下出现的以肺泡内、肺泡管、呼吸性细支气管及终末细支气管腔内有机化性肉芽组织为病理特点,对糖皮质激素治疗反应良好的间质性肺疾病。COP亚急性起病,病程多在2个月以内,发病前可出现流感样症状,最常见的临床症状为干咳和不同程度的呼吸困难,查体可在病变肺部位闻及爆裂音。COP的HRCT表现可为肺实变影或不规则线状、条索影沿支气管血管束分布,以及随机分布的磨玻璃影,病变呈游走性(图2)。肺活检病理是COP确诊的金标准。大部分COP对激素治疗反应好,临床症状及影像学可迅速改善,预后良好。
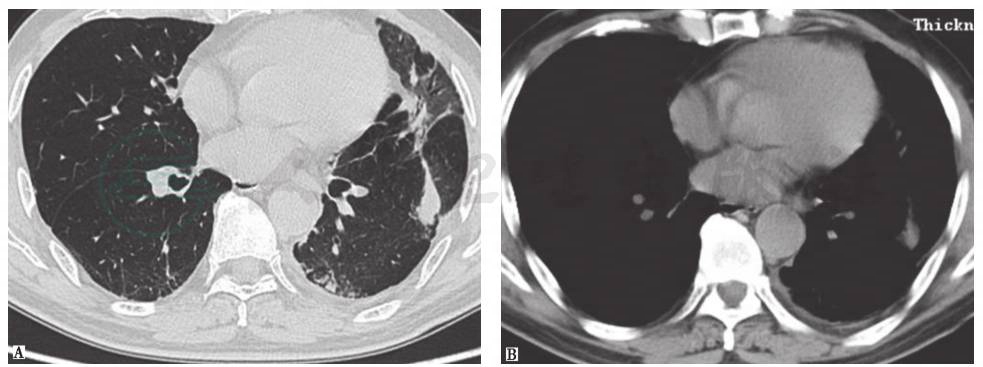
图2 隐源性机化性肺炎胸部CT表现
男性患者,66岁,咳嗽、气喘2个月余。胸部CT可见以左肺为主的磨玻璃高密度影伴实变病灶,沿支气管血管束走行分布,并见右下肺胸膜下间隔旁气肿。临床诊断为隐源性机化性肺炎
(3)脱屑性间质性肺炎(DIP)
是间质性肺炎的一种类型,是以气腔单核细胞浸润为特征的慢性肺部炎症。病变的主要部位在细支气管及周围的气腔,其发病机制与外源性致病因子吸入和吸烟关系密切,因此与呼吸性细支气管炎伴间质性肺疾病及肺朗格汉斯细胞组织细胞增生症一起归为吸烟相关的间质性肺疾病。DIP的病理学特征为肺泡腔弥漫性分布均一的肺泡巨噬细胞。最常见的临床表现为进行性加重的活动后气促及呼吸困难,查体双下肺有吸气末Velcro音。支气管肺泡灌洗液(BALF)中可见各类细胞(中性粒细胞、嗜酸性粒细胞、淋巴细胞,尤其是肺泡巨噬细胞)总数明显增多。BALF中见大量褐色素性肺泡巨噬细胞可协助诊断。典型HRCT表现为以中下肺为主的磨玻璃影,随着病情进一步发展,可表现为肺底部和胸膜下网格影、不规则条索影,结节影和蜂窝影比较少见(图3)。DIP的治疗主要是立即戒烟(部分患者戒烟后病情可自行缓解),其次是尽早应用糖皮质激素。
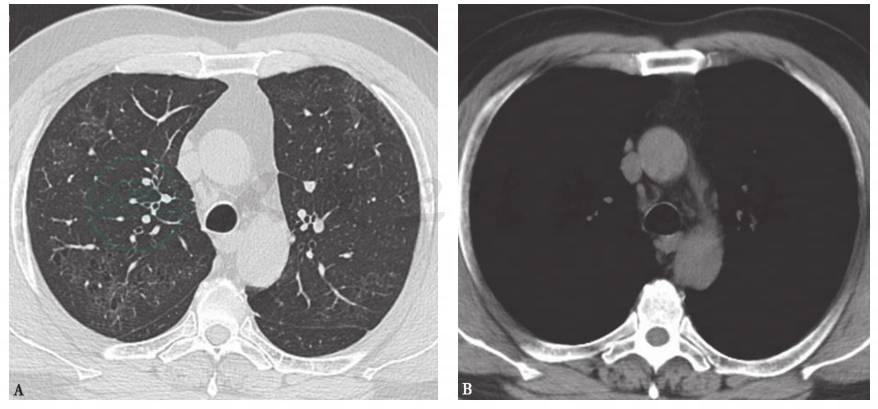
图3 DIP胸部CT表现
男性患者,59岁,吸烟,咳嗽2个月余,临床诊断为DIP。胸部CT可见双肺弥漫性磨玻璃影
(4)结缔组织疾病相关间质性肺疾病
结缔组织疾病(CTD)是一组异质性、免疫介导、可累及多个器官的炎症性病变,主要包括类风湿关节炎(rheumatoid arthritis,RA)、系统性红斑狼疮(systemic lupus erythematosus,SLE)、 多发性肌炎(polymyositis,PM)/皮肌炎(dermatomyositis,DM)、系统性硬化症(systemic sclerosis,SSc)、干燥综合征(Sjogren syndrome,SS)等。CTD的主要病理改变为疏松结缔组织发生黏液性水肿、类纤维蛋白变性、小血管坏死和组织损伤。肺支气管、肺血管及肺间质及胸膜含有丰富的结缔组织,因而成为重要的靶器官。CTD累及肺的表现有间质性肺疾病、细支气管炎、肺血管病变及肺实质小结节等。病理类型可有普通型间质性肺炎、非特异性间质性肺炎、淋巴细胞间质性肺炎、机化性肺炎等。不同的病理类型有不同的HRCT表现(图4)。临床表现不仅有原发疾病表现也有肺部表现,诊断需要完善自身免疫相关疾病的诊断,必要时需要肺活检明确组织病理学类型,与对治疗的反应和预后关系比较密切。

图4 皮肌炎合并机化性肺炎胸部CT表现
女性患者,46岁,活动后气喘1个月。胸部CT可见双肺磨玻璃高密度影伴实变病灶,沿支气管血管束走行分布。病理诊断为皮肌炎合并机化性肺炎
(5)放射性肺炎
是一种剂量依赖性肺损伤。研究显示,放射性肺炎在照射剂量低于20Gy时很少发生,而通常发生在照射剂量高于60Gy时。除了照射剂量,放射性肺炎的发生还与分割方式、照射野范围等治疗因素,以及患者的基础肺病、放疗暴露史等宿主条件有关。放射性肺炎的发病时间窗一般在放疗结束后4周~6个月,而急性放射性肺炎通常发生在放疗结束后4~12周。主要临床症状包括发热、咳嗽、气短,患侧肺部听诊可闻湿啰音。放射性肺炎的病理特点在急性期为肺泡炎,慢性期为纤维化。胸部X线征象为照射野同侧肺野出现磨玻璃样渗出或实变,可合并胸腔积液;有时对侧肺野也可出现病变(图5)。胸部HRCT可观察到照射野同侧肺野弥漫性磨玻璃样渗出或实变、小叶间隔及小叶内间隔增厚;慢性期可呈纤维化改变,包括牵拉性支气管扩张及蜂窝样改变。
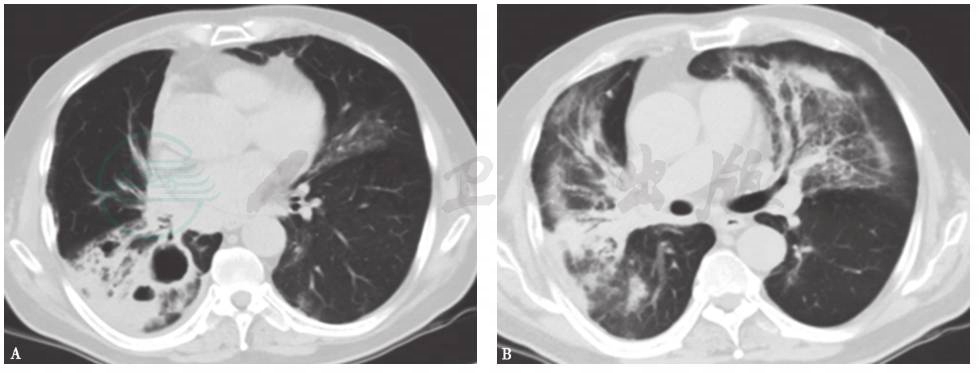
图5 放射性肺炎胸部影像学表现
男性患者,59岁,右下肺鳞癌放疗结束后6周(48Gy)出现双肺弥漫渗出。HRCT见右下叶癌性空洞,空洞周围实变(A),双肺磨玻璃样渗出(B)
本例患者病程为亚急性,在过去的2个月内病情相对稳定(既无恶化也无好转);临床表现为活动后胸闷、气喘,伴有肢体活动受限及乏力感;既往史及个人史无特殊;无特殊的用药史;外院胸部CT提示双肺间质性改变。综合以上特点,考虑为特发性肺纤维化、脱屑性间质性肺炎、过敏性肺炎可能性不大。因患者有肢体活动受限,查体有双下肺捻发音及关节部位的皮疹脱屑,为CTD相关ILD可能性较大,但尚需要完善血常规、肿瘤指标物以及自身抗体、ANCA等免疫相关检查。
患者入院前2个月已经过正规抗感染治疗,疗程足,治疗后咳嗽、咳痰症状有改善,但活动后胸闷、气喘症状无明显改善。患者系中年女性,胸部CT表现为胸膜下磨玻璃影及斑片状实变影,目前主要诊断集中于结缔组织疾病相关肺间质性肺疾病,需要完善自身免疫病相关检查以明确具体疾病分类。
(一)临床信息
【实验室检查】
血常规:WBC 4.2×109/L,N% 48.1%,L% 35.2%,Hb 122g/l,PLT 271×109/L。
尿、便常规正常。
ESR 28mm/1h,CRP及结核抗体阴性。
生化:总蛋白(TP)55.7g/L,白蛋白(ALB)32.9g/L,甲状腺球蛋白(thyroglobulin,TG)2.61mmol/L。
自身抗体:抗SSA/Ro60KD抗体、抗SSA/Ro52KD抗体和抗Jo-1均阳性,抗角蛋白抗体及抗环瓜氨酸肽抗体阴性。
痰细菌培养阴性,真菌培养阴性;3次痰抗酸杆菌染色阴性;巨细胞病毒DNA、EB病毒DNA均未见异常。
心肌酶:乳酸脱氢酶(LDH)316U/L,肌酸激酶(CK)510U/L,心肌型肌酸激酶同工酶(CK-MB)23U/L,α-羟丁酸脱氢酶(α-hydroxybutyrate dehydrogenase,α-HBDH)254.7U/L。
血气分析:PaO2 58mmHg,其他基本正常。
肿瘤标志物:CEA 0.93ng/ml,CA19-9 17.35U/ml,AFP 2.86ng/ml,CA15-3 9.06U/ml,CA125 57.97U/ml,NSE 14.30ng/ml。
【肺功能检查】
以限制为主的中度混合性通气功能减退,弥散功能重度降低(VC 59.8%预计值,FVC 61.6%预计值,FEV1 61.6%预计值,FEV1%/FVC 78.01%,MVV 57.2% 预计值)。
【影像学检查】
腮腺ECT:双侧腮腺分泌及排泄功能正常。
心脏彩超:三尖瓣轻-中度、二尖瓣轻度反流,左室舒张功能减退,微量心包积液,射血分数(ejection fraction,EF)60%,肺动脉收缩压30mmHg。
胸部HRCT:入院后复查胸部HRCT见双肺胸膜下磨玻璃影及斑片状实变影,双侧可见胸膜下线(图6)。
双手正位X线片:未见明显骨性异常(图7)。
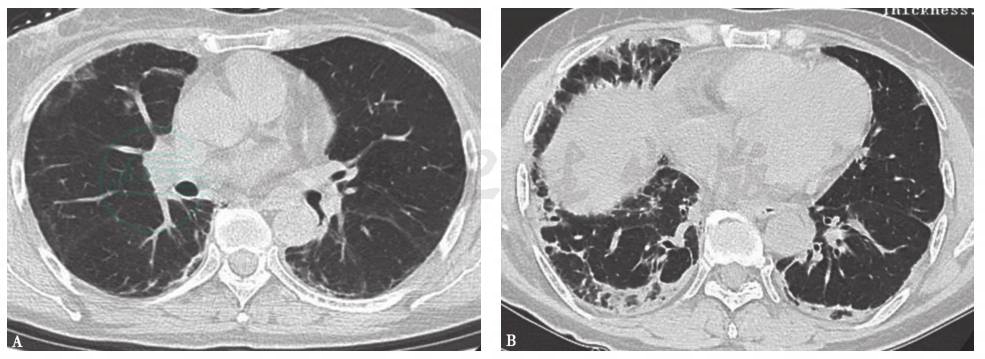
图6 入院后胸部HRCT表现

图7 双手正位X线片
【其他检查】
眼部检查:泪流量:左10mm/s,右7mm/s;双侧角膜荧光染色未见异常。
肌电图检查:未见明显异常。
唇腺黏膜活检:结果符合慢性炎症细胞浸润Ⅱ级。
(二)临床思辨
综合分析患者入院后的系列检查结果:①血常规基本正常,无嗜酸性粒细胞增高,故基本可排除过敏性肺炎、慢性嗜酸性粒细胞肺炎;②血气分析提示低氧血症,肺功能检查提示有弥散功能障碍,符合间质性肺疾病改变;③多次痰培养、抗酸涂片、病毒等相关病原学检查阴性,故感染性疾病可能性不大;④自身抗体SSA阳性、抗Jo-1抗体阳性,唇腺黏膜活检符合慢性炎症细胞浸润Ⅱ级,肌酶谱增高,结合患者临床表现,诊断倾向于皮肌炎和干燥综合征。
(一)临床信息
本例患者为中年女性,症状表现为肢体活动受限,查体见双肘关节皮肤干燥、脱屑,肌酶升高,支持皮肌炎的诊断。患者同时出现呼吸系统受累表现(活动后憋气),胸部CT示双肺间质病变,血清抗Jo-1抗体阳性,故抗Jo-1抗体综合征的诊断明确(抗Jo-1抗体综合征是一组临床表现为肺间质病变、对称性多关节炎、雷诺现象、技工手等,且血清抗Jo-1抗体阳性的症候群)。
最后诊断:抗Jo-1抗体综合征合并干燥综合征伴间质性肺炎。
诊断明确后,给予甲泼尼龙(40mg,每天2次)、羟氯喹(100mg,每天2次)、环磷酰胺(0.4g/2w)免疫抑制治疗,症状好转后序贯改为口服泼尼松(15mg,每天1次)和羟氯喹(0.2g,每天2次)治疗1周后,患者咳嗽、咳痰症状较前明显好转,双肩关节疼痛及下蹲后站起乏力明显缓解,考虑治疗有效。患者病情稳定出院,继续口服药物治疗。1个月后,泼尼松减量(10mg,每天2次)维持治疗,羟氯喹(0.2g,每天2次)及环磷酰胺0.4g/2周。6个月后复查胸部HRCT,见肺内病变较前有明显吸收(图8)。
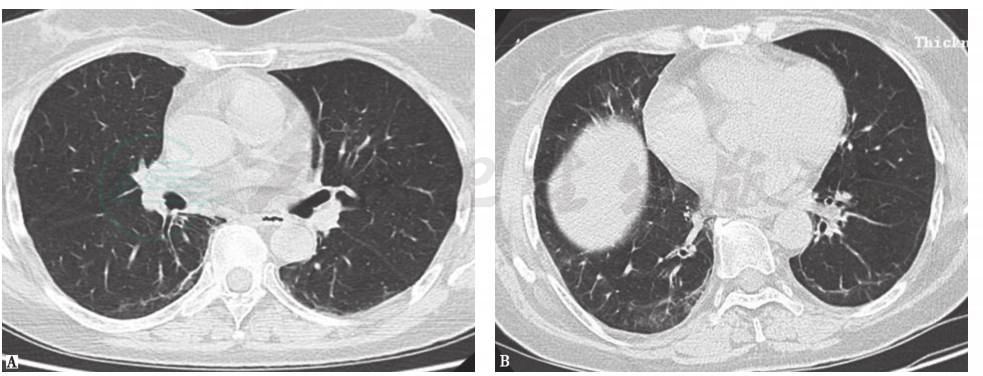
图8 6个月后复查胸部HRCT表现
HRCT示双肺胸膜下病变较半年前明显吸收,双侧胸膜下线可见
(二)临床思辨
本病例经临床多个侧面甄别获得确诊。治疗随访复查胸部CT示病灶较前明显吸收(图8),病情稳定。据有文献报道,50%多发性肌炎/皮肌炎(PM/DM)患者合并有间质性肺疾病。
抗组氨酰抗体(以抗Jo-1抗体为主)是多发性肌炎及皮肌炎的特异性抗体。国外许多报告表明,此抗体阳性的患者具有一组特殊的症候群,即肺间质病变、对称性多关节炎、雷诺现象、技工手等,称抗Jo-1抗体综合征。抗Jo-1抗体阳性对诊断较为特异,应予以重视。
抗Jo-1抗体临床意义:在不明原因间质性肺疾病鉴别中,须注意抗Jo-1抗体综合征。肌炎可能在间质性肺疾病发病多年后再出现,在没出现肌炎的典型症状前,抗Jo-1抗体可协助诊断。抗合成酶抗体阳性常提示预后不良。值得注意的是,由于抗Jo-1抗体的靶抗原是组氨酰tRNA合成酶,主要存在于细胞质内,而抗核抗体检测的主要是细胞核内抗原,故抗核抗体阴性并不等于抗Jo-1抗体亦阴性。因此,临床上对于高度怀疑抗Jo-1抗体综合征的患者,应积极行抗Jo-1抗体检测,必要时予以重复检测。这类患者治疗效果差,预后差,撤药后病情易复发,尽早诊疗则可以提高预期寿命,减少病死率。
抗Jo-1抗体综合征诊断:抗Jo-1抗体综合征的临床表现多样,包括多发性肌炎或皮肌炎、肺间质病变、多关节炎、雷诺现象、技工手,指(趾)过度硬化,面部毛细血管扩张,钙化及干燥症等,同时可有发热、全身乏力、体重减轻等全身表现。多项临床研究及病例报道均提示,在抗Jo-1抗体综合征患者中,炎性肌病相关症状很常见,但表现往往轻微,甚至常不作为首发症状而易被临床医师忽略,个别患者可无肌肉受累表现。肌肉受累主要表现为肌痛、肌无力、肌肉萎缩及纤维化等,首先累及四肢近端肌肉,检查可发现血清肌酶升高、肌电图变化及肌活检异常等。肺间质病变甚至可以是此病唯一的临床表现,主要症状为胸闷、干咳及呼吸困难,一些患者可出现急性呼吸窘迫综合征。抗Jo-1抗体综合征可根据肺部表现的发生及发展分为急性进展型、慢性迁延型及无症状型,仅少数患者表现为急性进展型。肺功能检查结果显示限制性通气功能障碍。急性发病患者的肺部HRCT表现为双肺(肺底部为著)网格和磨玻璃影,蜂窝影和牵拉性支气管扩张少见。肺组织活检可为此病的诊断和预后判断提供更为可靠的依据。其主要病理类型可分为机化性肺炎(OP)、非特异性间质性肺炎(NSIP)、普通型间质性肺炎(UIP)。OP常见于急性进展型患者,NSIP及UIP常见于慢性迁延型患者。
抗Jo-1抗体综合征治疗:尚无标准治疗方案,首选药物为糖皮质激素,可改善患者关节、肌肉及全身症状,对部分肺间质病变也有一定效果。在肺间质病变中,OP和NSIP对糖皮质激素的治疗反应较好,UIP则常表现为激素抵抗,需其他免疫抑制剂治疗。激素治疗效果不佳者应尽早使用其他免疫抑制剂,以防出现不可逆的肺部损伤而影响预后,例如可以联合秋水仙碱、甲氨蝶呤、环磷酰胺、硫唑嘌呤、环孢霉素A、霉酚酸酯、他克莫司等免疫抑制剂,难治性病例可应用免疫球蛋白、细胞因子抑制剂(如IL-1抑制剂、TNF-α抑制剂、转化生长因子β抑制剂)以及利妥昔单抗等。已经证实的是,在肺组织形成不可逆肺泡-毛细血管壁损伤之前尽早控制肺泡炎是治疗成功的关键,联合使用免疫抑制剂的作用优于单一激素治疗。
咳嗽、气喘是弥漫性间质性肺病最常见的临床表现,无特异性,而胸部CT呈磨玻璃影可见于感染性疾病(如肺结核、肺孢子菌肺炎及病毒性肺炎),亦可见于一些间质性肺疾病。这些疾病虽然有相似的临床症状和影像学改变,但在宿主条件、危险因素、临床症状、实验室检查、影像学表现、治疗反应等方面又各有独特表现,而这些独特性正是鉴别诊断的关键。在临床工作中,对于原因不明的女性肺间质病变患者,应注意询问是否伴有关节肿痛、肌无力、雷诺现象、皮疹等结缔组织疾病表现,细致体检,并完善肌酶谱、肌电图检查,抗核抗体(尤其是抗Jo-1抗体)检测等,及时诊断,及早治疗,以改善预后,降低病死率。
(马苗 蔡后荣)
1.Erasmus JJ,Bucci MK,Munden RF.Radiation-induced lung disease.Imaging of the Chest. US:Saunders,2008,82:1225-1240.
2.邓文静,张秋红,张玉萍.以肺部为主要表现的抗Jo-1抗体综合征1例及文献复习.中华肺部疾病杂志(电子版),2015,8(1):54-57.
3.关岚,彭丽滢,李萌.抗合成酶抗体综合征合并肺间质病1例并文献复习.中国医刊,2014,49(10):28-30.
4.Hironao Hozumi,Noriyuki Enomoto,Masato Kono.Prognostic significance of antiaminoacyl-tRNA synthetase antibodies in polymyositis/dermatomyositis-associated interstitial lung disease :a retrospective case control study.PLoS ONE,2014,10(3):e0120313.
5.Ju Sun Song,Jiwon Hwang,Hoon-Suk Cha.Significance of myositis autoantibody in patients with idiopathic interstitial lung disease.Yonsei Med J,2015,56(3):676-683.
6.Thomas J,Richards,Aaron Eggebeen.Characterization and peripheral blood biomarker assessment of Jo-1 antibody-positive interstitial lung disease.Arthritis Rheum,2009,60(7):2183-2192.
7.Hidenaga Kawasumi,Takahisa Gono,Yasushi Kawaguchi.Recent treatment of interstitial lung disease with idiopathic inflammatory myopathies.Clinical Medicine sights :Circulatory,Respiratory and Pulmonary Medicine,2015,9(1):9-17.
8.Marie I,Dominique S,Janvresse A.Rituximab therapy for refractory interstitial lung disease related to antisynthetase syndrome.Respiratory Medicine,2012,106(4):58l-587.









